使用python来分析一下张泽民院士2018年的单细胞数据
- 2026-06-27 11:19:35
python基本部分学完了之后,见专辑《python生信笔记》,就需要开始进行大量的数据分析实战练习进行掌握了。今天来学习一篇顶刊杂志的单细胞数据处理,使用python!
note:这个数据主要来自最近开一个直播进行实战训练,详细情况以及进群方式见:python单细胞数据分析实战直播交流群
直播计划大纲:https://www.yuque.com/xinnigegui-uqv7c/nneog4/ecg5ec5hscuiudqq
数据背景
Guo_Zhang_2018_NSCLC 数据处理
今天分析直播的那篇文献里面里面提到的 Guo, Zhang et al. 数据,来自文献 10.1038/s41591-018-0045-3 。文献标题为《Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing》。 2018 年6月25号发表在杂志Nature Medicine。
数据集为:data/12_input_adatas/guo_zhang_2018_nsclc.h5ad,在文献里面的下载地址为 :https://zenodo.org/records/7227571,在里面的 input_data.tar.xz
文献内容简介
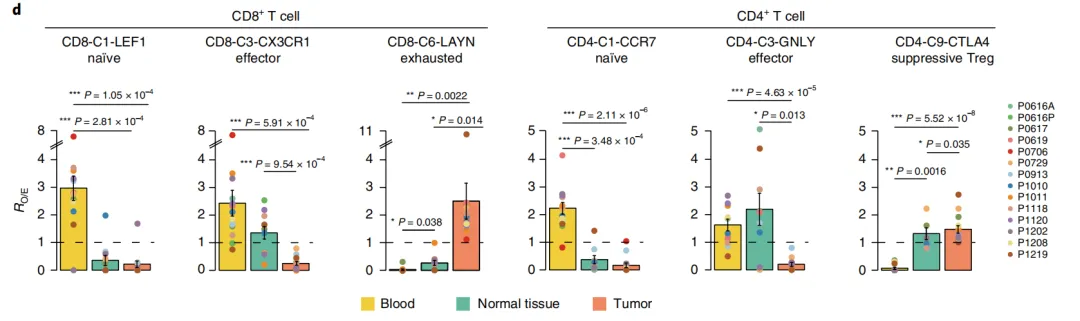
本研究对 14 例未经治疗的非小细胞肺癌患者的 12,346 个T细胞进行了深度单细胞RNA测序。通过联合基因表达谱与基于T细胞抗原受体的谱系追踪技术,发现了具有高迁移特性的组织间效应T细胞的重要亚群。除处于耗竭状态的 tumor-infiltrating CD8+ T cells 外,还观察到两个处于耗竭前状态的细胞群,且 pre-exhausted 与 exhausted T cells 的高比例与肺腺癌患者的良好预后相关。此外,研究还发现 the tumor regulatory T cells (Tregs) 存在更深层异质性,其特征表现为抗原特异性 Tregs 激活标志物 TNFRSF9 的双峰分布。这些活化肿瘤 Tregs 的基因标志(包括 IL1R2 )与肺腺癌不良预后相关。
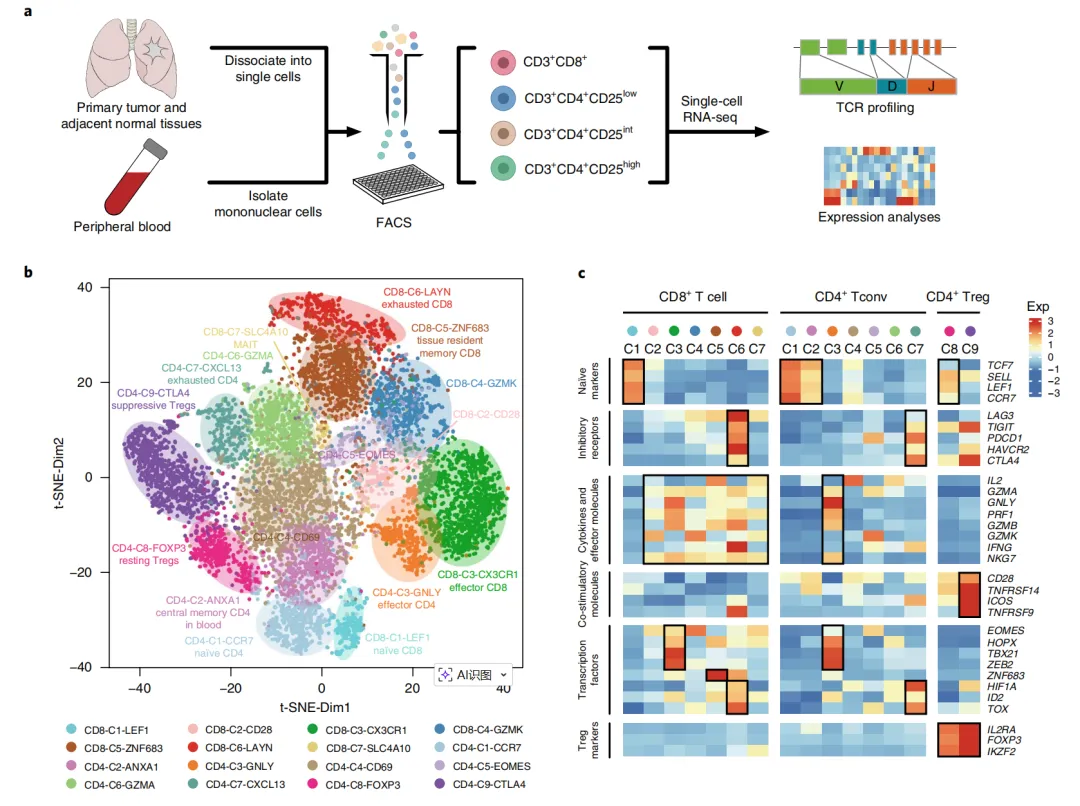
细胞类型比例在不同分组中的比例差异:
数据读取
先加载模块:
# %%# %load_ext autoreload# %autoreload 2import scanpy as scimport numpy as npimport pandas as pdimport scipy.sparse as spfrom scanpy_helpers.annotation import AnnotationHelperfrom nxfvars import nxfvarssc.set_figure_params(figsize=(5, 5))# %%adata = sc.read_h5ad(filename="data/12_input_adatas/guo_zhang_2018_nsclc.h5ad" )adata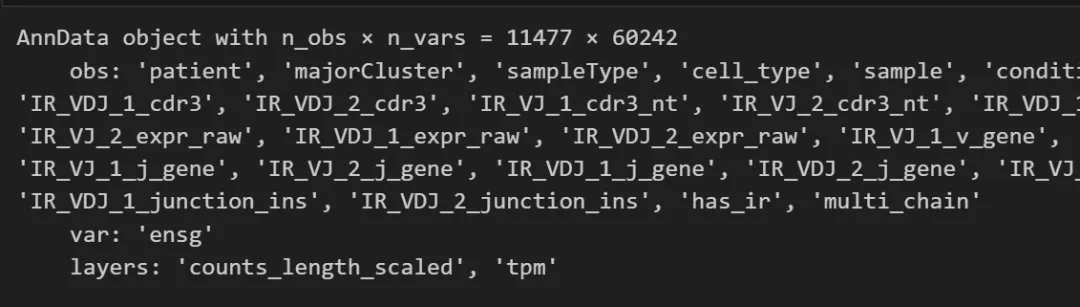
11477个细胞。
其他信息
看看给的数据里面其他的表型:
adata.obsadata.obs['patient'].value_counts()adata.obs['condition'].value_counts()adata.obs['origin'].value_counts()adata.obs['majorCluster'].value_counts()adata.obs['cell_type'].value_counts()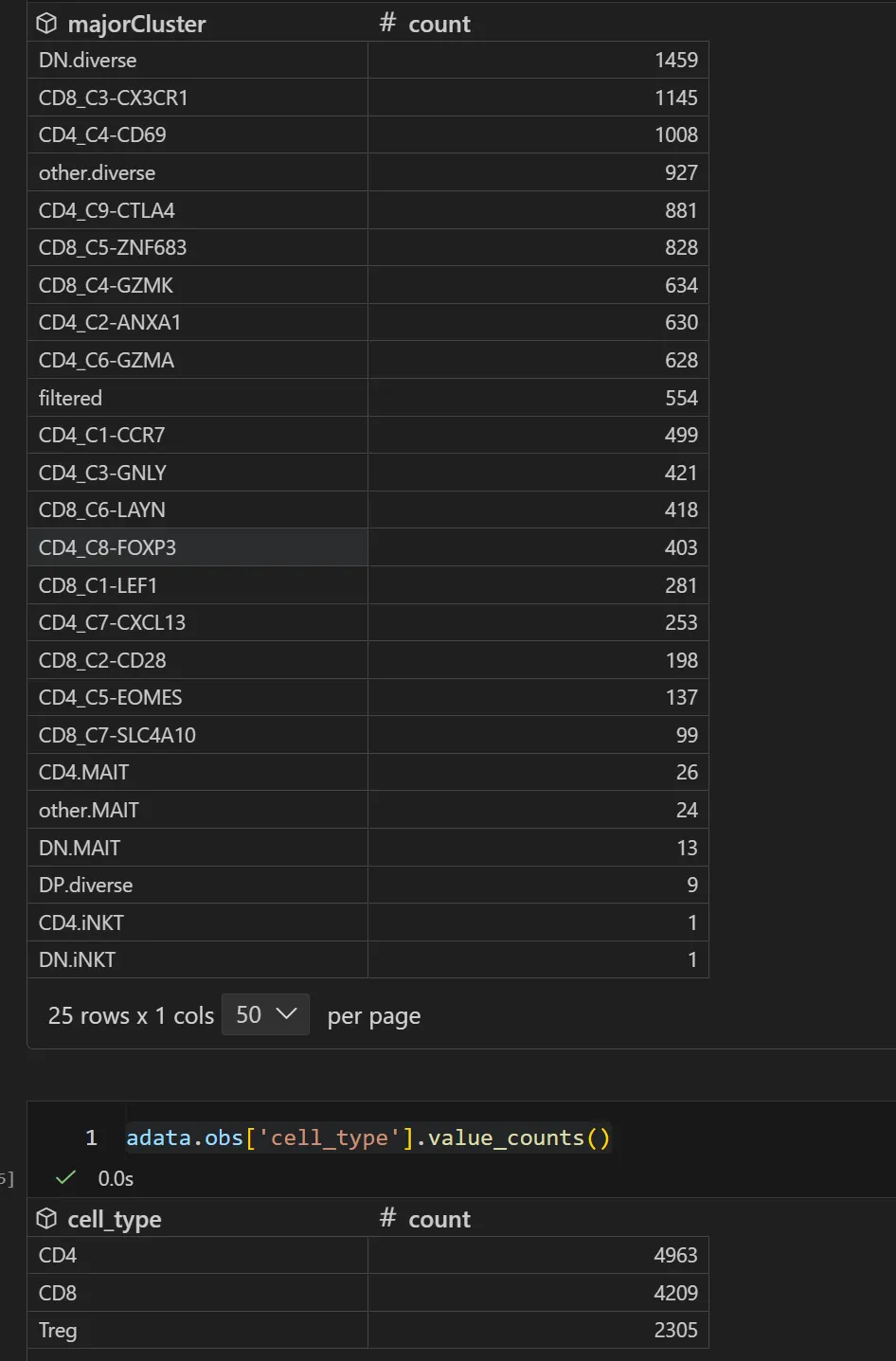
细胞给了详细的注释。
数据过滤
我们目前已经做了好几个数据的python代码训练,所以这里应该很熟悉。
计算每个细胞中线粒体基因,核糖体基因,红血细胞基因表达比例:
# 计算线粒体基因比例adata.var['mt']=adata.var_names.str.startswith('MT-')# 计算核糖体基因比例adata.var['ribo']=adata.var_names.str.match('^RP[SL]')# 计算红血细胞基因比例adata.var['hb']=adata.var_names.str.match('^HB[^(P)]')# 计算sc.pp.calculate_qc_metrics(adata,qc_vars=['mt','ribo','hb'],percent_top=None,log1p=False,inplace=True)# 质控adata.obs.head()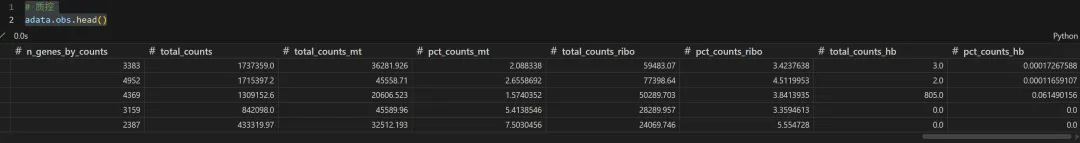
计算结果,然后可视化看看:
from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (9, 10)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="patient", jitter=0.4,rotation=90,multi_panel=True, )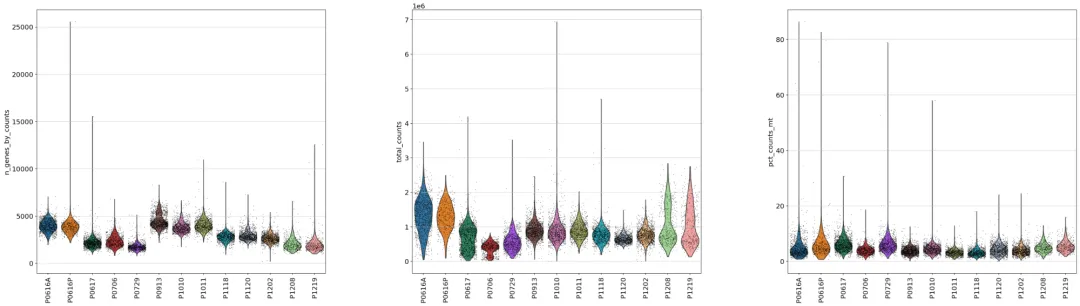
看看文献给的过滤标准:
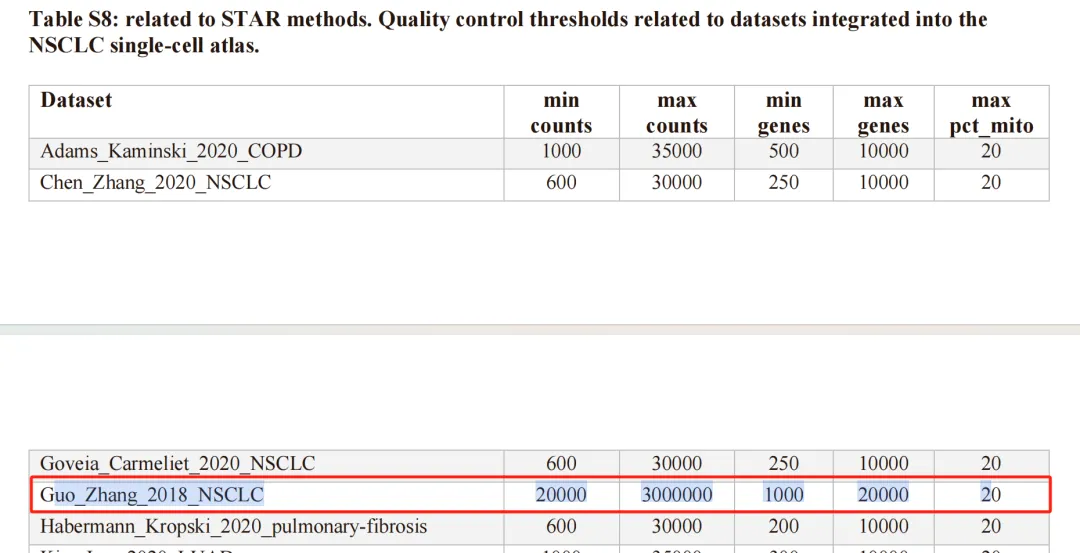
过滤:
## 过滤前的数据adata.shape# 基本过滤sc.pp.filter_genes(adata, min_cells=3)## 过滤# 方法2:一次性过滤所有条件(更高效)adata = adata[ (adata.obs['n_genes_by_counts'] > 1000) & (adata.obs['n_genes_by_counts'] < 20000) & (adata.obs['total_counts'] > 20000) & (adata.obs['total_counts'] < 3000000) & (adata.obs['pct_counts_mt'] < 20), :].copy()## 过滤后的数据adata.shape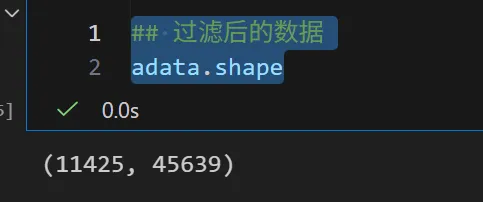
过滤前后细胞数没有很大变化。
过滤后的小提琴:
from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (9, 10)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="patient", jitter=0.4,rotation=90,multi_panel=True,#stripplot=False, # remove the internal dots#inner="box", # adds a boxplot inside violins )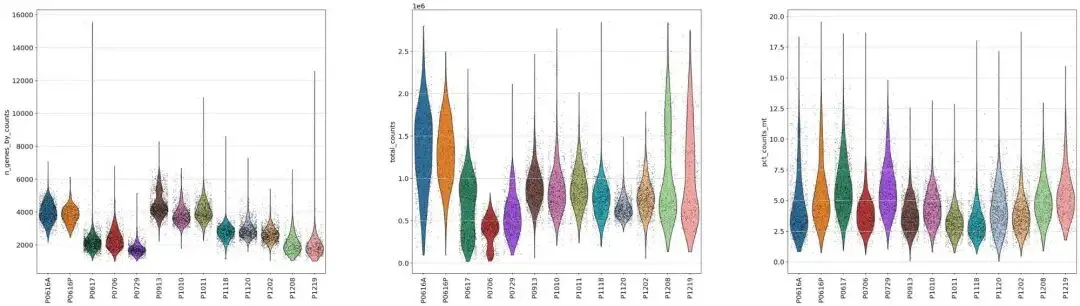
保存数据:
## 保存数据adata.write("01_qc/guo_zhang_2018_nsclc_qc.h5ad")降维聚类
scanpy的标准流程:
# 首先将数据矩阵标准化(校正文库大小):sc.pp.normalize_total(adata,target_sum=1e4)# 对数据进行logsc.pp.log1p(adata)adata.raw = adata.copy()# 高变基因sc.pp.highly_variable_genes(adata)# 归一化sc.pp.scale(adata)# pca分析sc.tl.pca(adata,use_highly_variable=True)# harmony整合sc.external.pp.harmony_integrate(adata,'patient')# 聚类sc.pp.neighbors(adata,use_rep='X_pca_harmony',n_pcs=30)# sc.pp.neighbors(adata,use_rep='X_pca',n_pcs=30)sc.tl.umap(adata)# Using the igraph implementation and a fixed number of iterations can be significantly faster, especially for larger datasetssc.tl.leiden(adata, flavor="igraph", n_iterations=2,resolution=0.5)可视化一下:
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (6.5, 6)}): sc.pl.umap(adata, color=["leiden",'patient'], legend_loc="on data", size=7)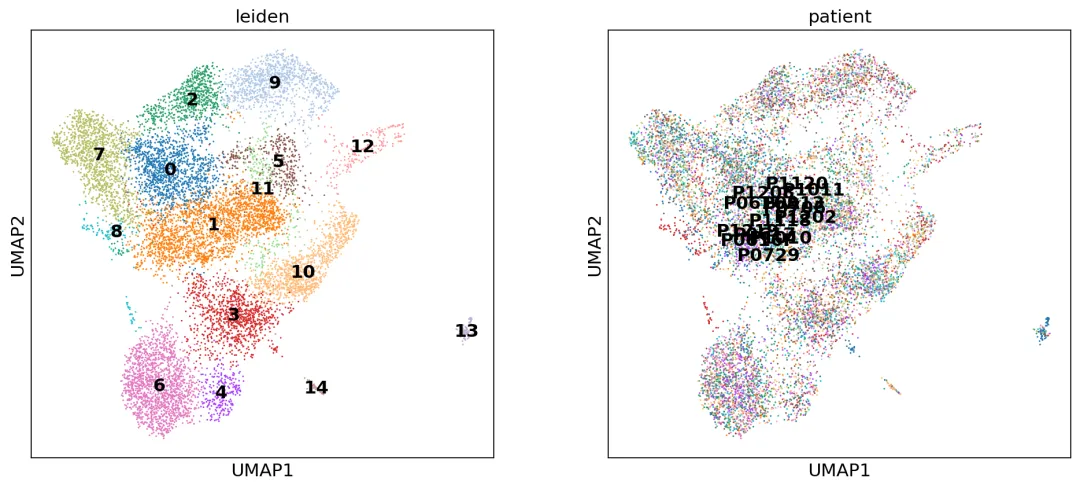
harmony整合效果还可以。但是我打算后面试一下其他的整合方法。
看看亚群
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (8, 7)}): sc.pl.umap(adata, color=["cell_type"], legend_loc="on data", size=7) # 样本太多了所以是灰色?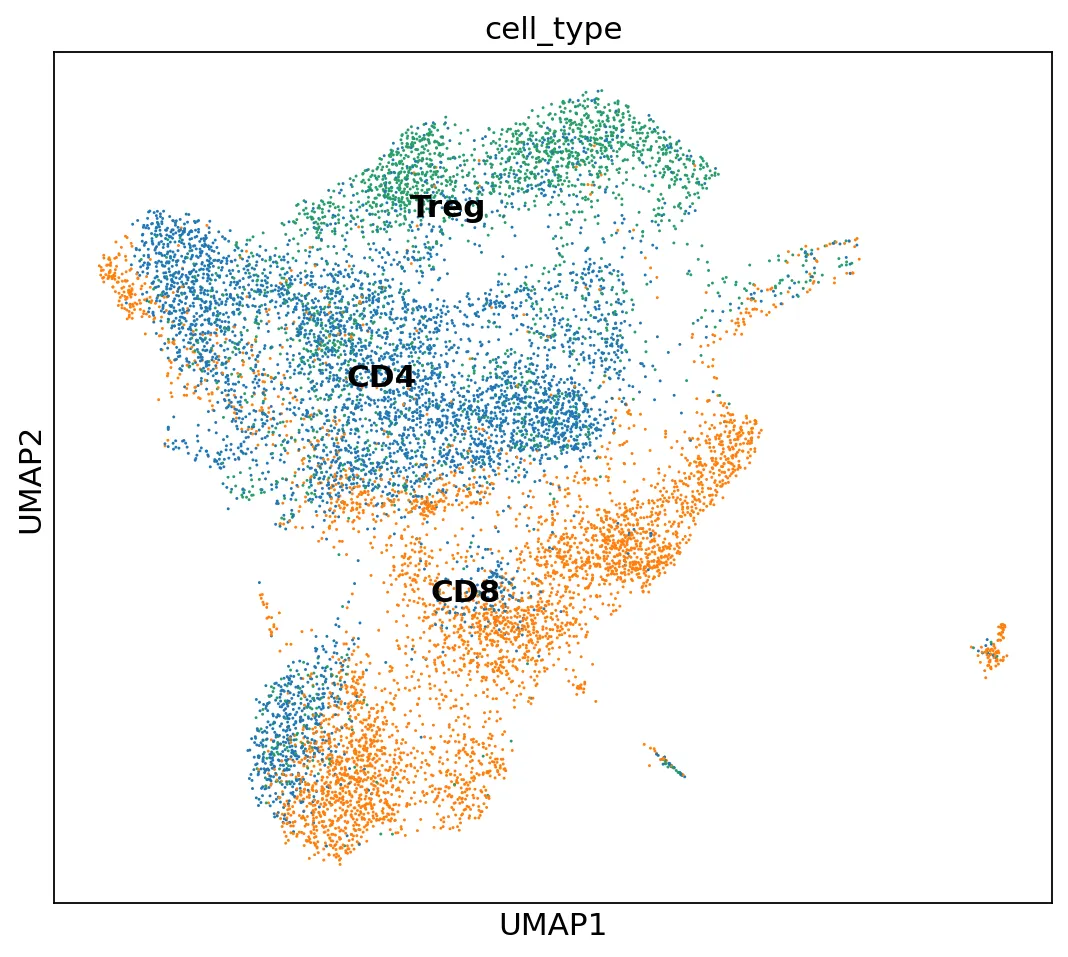
跟原有的注释结果还算吻合,CD8与CD4有比较清晰的分群界限。
看看marker基因
sc.pl.umap(adata, color=["PTPRC", "CD3D","CD8A","CD8B","CD4","NKG7"])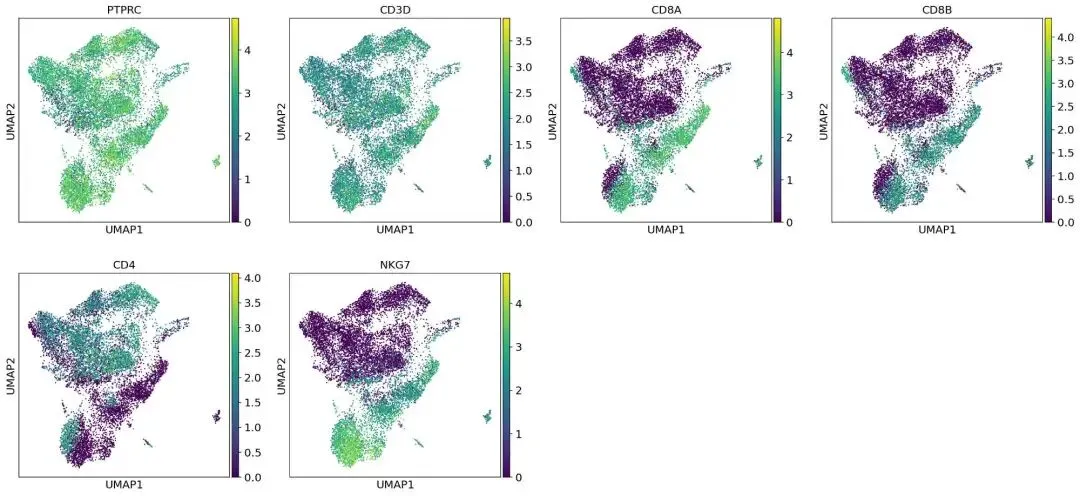
最后保存一下数据:
## 保存数据adata.write("01_qc/guo_zhang_2018_nsclc_umap.h5ad")今天分享到这里,要一起来学习python单细胞数据分析吗,加我Biotree123,进python分析群一起实战~
转发:
生信入门&数据挖掘线上直播课2026年1月班,你的生物信息学入门课 时隔5年,我们的生信技能树VIP学徒继续招生啦 满足你生信分析计算需求的低价解决方案 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑

