编者按
我们将推出一个新的栏目,重构算法的介绍,在这个栏目中,我们将介绍omicverse开源重构生态最新的成果以及完整的教程,欢迎关注。
本期教程里,我们给大家带来的是BCR免疫组库分析:这是omicverse中 ov.airr 模块的内容。我们在omicverse中整合了BCR分析里常用的一套流程,包括免疫球蛋白基因使用、B细胞克隆推断、克隆扩增、多样性、germline重建、体细胞高频突变、BASELINe抗原选择、谱系树、IGHV基因型推断以及isotype switching分析。
如果说TCR分析更常问“这个T细胞可能识别什么抗原”,那么BCR分析很多时候问的是另一件事:一个B细胞克隆在抗原刺激后,是否真的经历了扩增、突变、选择和换类。
本文使用Laserson 2014流感疫苗接种前后IgH数据,演示如何用OmicVerse重建一次B细胞亲和力成熟过程,希望本期教程对你有所帮助。
本文AI率20%,阅读大约需要6min。
OmicVerse团队
1. 引言
BCR和TCR都是免疫受体,但二者的分析重点完全不一样。
T细胞的TCR在胸腺里形成后,通常不会在外周免疫反应中继续突变。因此TCR分析更常关注克隆扩增、抗原特异性、public clonotype、TCRdist或者GLIPH这类相似性分析。
B细胞不一样。
B细胞遇到抗原后,会进入生发中心。在那里,BCR会经历一套很经典的过程:
- 第二,AID酶在IgH可变区引入体细胞高频突变,也就是SHM。
- 第四,一部分B细胞从IgM/IgD换类到IgG/IgA。
所以BCR repertoire不是一张普通的克隆型表,它更像是一份免疫反应的历史记录。
如果我们拿到疫苗接种前和接种后第7天的IgH序列,就可以问很多很具体的问题:
- 突变是不是更多落在CDR区域,而不是FWR结构区域?
- IgG/IgA这类class-switched细胞是否明显增加?
本期教程就用OmicVerse把这一整套流程跑一遍。
2. 数据读入
BCR这部分分析用的是AIRR格式的 DataFrame,不是AnnData结构。每一行是一条重排后的免疫球蛋白重链序列。
首先载入OmicVerse:
import omicverse as ov
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
ov.plot_set()
读取Laserson 2014流感疫苗数据:
bcr = ov.datasets.airr_bcr()
print(f"sequences: {bcr.shape[0]} columns: {bcr.shape[1]}")
print(f"timepoints: {bcr['sample_id'].value_counts().to_dict()}")
print(f"isotypes: {bcr['c_call'].value_counts().to_dict()}")
sequences: 1999 columns: 24
timepoints: {'-1h': 1000, '+7d': 999}
isotypes: {'IGHM': 718, 'IGHG': 650, 'IGHA': 372, 'IGHD': 259}
这个数据一共有1999条IgH序列,分成两个时间点:
然后只保留productive rearrangements,也就是功能性、读码框正确、没有提前终止密码子的重排序列。
db = bcr[bcr['productive'] == 'T'].copy()
print(f"productive sequences: {len(db)} ({len(bcr) - len(db)} non-productive dropped)")
iso = ov.airr.isotype_class(db)
print(iso.value_counts().to_dict())
productive sequences: 1780 (219 non-productive dropped)
{'naive (IgM/IgD)': 922, 'switched (IgG/IgA)': 858}
也就是说,这个数据里大约一半是naive相关的IgM/IgD,另一半是class-switched的IgG/IgA。
这很适合讲B细胞疫苗反应。因为疫苗后真正进入生发中心的B细胞,往往会同时表现出三个特征:扩增、SHM升高、isotype switching。
3. 免疫球蛋白基因使用
在做克隆和突变之前,我们先看IgH使用了哪些V/J基因。
usage_v = ov.airr.bcr_gene_usage(db, gene='v_call', mode='family')
usage_v = usage_v.sort_values('seq_freq', ascending=False)
print("IGHV family usage across the productive repertoire:")
print(usage_v[['gene', 'seq_count', 'seq_freq']].to_string(index=False))
IGHV family usage across the productive repertoire:
gene seq_count seq_freq
IGHV3 1118 0.628090
IGHV4 265 0.148876
IGHV1 201 0.112921
IGHV2 95 0.053371
IGHV5 86 0.048315
IGHV6 15 0.008427
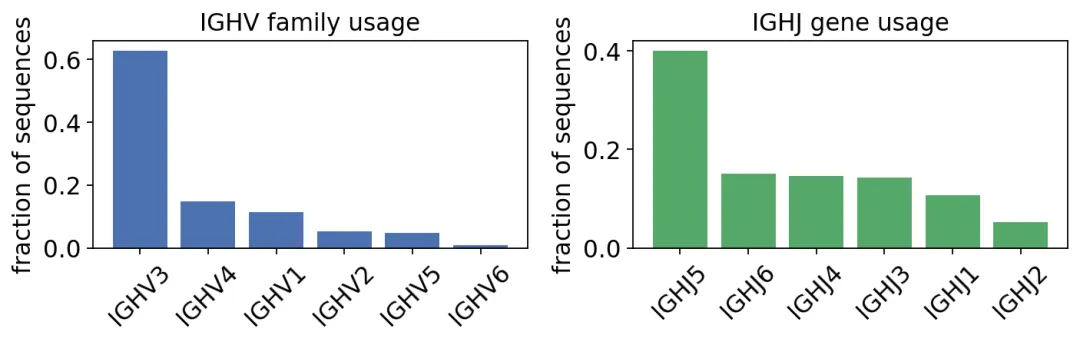 IGHV和IGHJ基因使用
IGHV和IGHJ基因使用这个结果很符合人类IgH repertoire的常见结构:IGHV3占比最高,IGHV4和IGHV1跟在后面。
然后我们按isotype拆开看:
pivot = ov.airr.bcr_gene_usage(
db,
gene='v_call',
mode='family',
groups='c_call',
pivot=True,
)
pivot = pivot.loc[['IGHV1', 'IGHV3', 'IGHV4', 'IGHV5']]
print("IGHV family fraction by isotype:")
print(pivot.round(3).to_string())
IGHV family fraction by isotype:
c_call IGHA IGHD IGHG IGHM
gene
IGHV1 0.060 0.194 0.020 0.184
IGHV3 0.768 0.463 0.899 0.403
IGHV4 0.119 0.227 0.067 0.201
IGHV5 0.033 0.062 0.013 0.079
可以看到,不同isotype的V gene使用比例并不完全一样。IgG和IgA中IGHV3占比更高,而IgM/IgD里IGHV1和IGHV4的比例更高。
4. B细胞克隆推断
BCR分析里的“克隆”和TCR克隆型不是同一件事。
TCR里,我们经常把完全相同的CDR3当成同一个克隆型。但B细胞经历SHM后,同一个祖先B细胞的后代会产生很多相似但不完全相同的序列。
所以BCR clone的定义更接近:它们来自同一个V(D)J重排祖先,V/J基因一致,junction长度一致,CDR3距离足够近。
首先用数据自己估计一个距离阈值:
threshold, db_dist = ov.airr.distance_threshold(
db,
model='ham',
first=True,
)
dist = db_dist['dist_nearest'].dropna()
print(f"inferred clonal threshold: {threshold:.3f}")
print(f"sequences with a same-VJL neighbour: {len(dist)}")
inferred clonal threshold: 0.168
sequences with a same-VJL neighbour: 1744
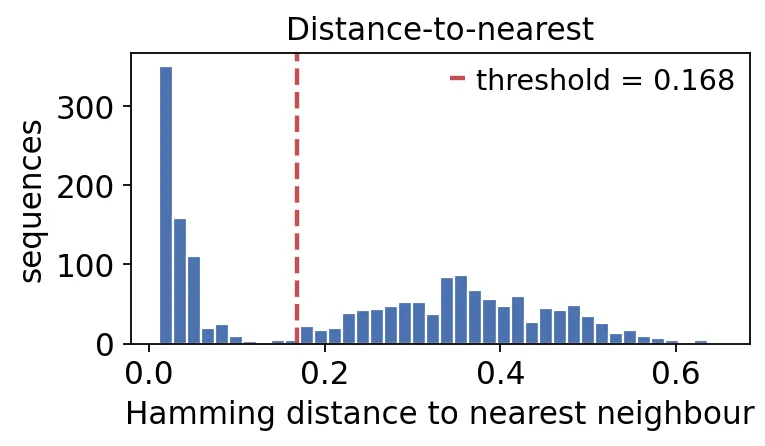 BCR克隆距离阈值
BCR克隆距离阈值这张图看的是每条序列到同V/J/长度邻居的最近距离。
低距离峰通常对应同一个B细胞克隆内部的序列,高距离峰对应无关重排。阈值放在两个峰之间,就可以把克隆内差异和克隆间差异分开。
然后做clonal clustering:
clones = ov.airr.clonal_clustering(
db.drop(columns=['clone_id']),
method='hierarchical',
threshold=threshold,
linkage='complete',
)
sizes = clones['clone_id'].value_counts()
print(f"B-cell clones inferred: {sizes.size}")
print(f" singletons: {(sizes == 1).sum()}")
print(f" expanded (>=3 seqs): {(sizes >= 3).sum()}")
print(f" largest clone: {sizes.max()} sequences")
B-cell clones inferred: 1018
singletons: 894
expanded (>=3 seqs): 49
largest clone: 165 sequences
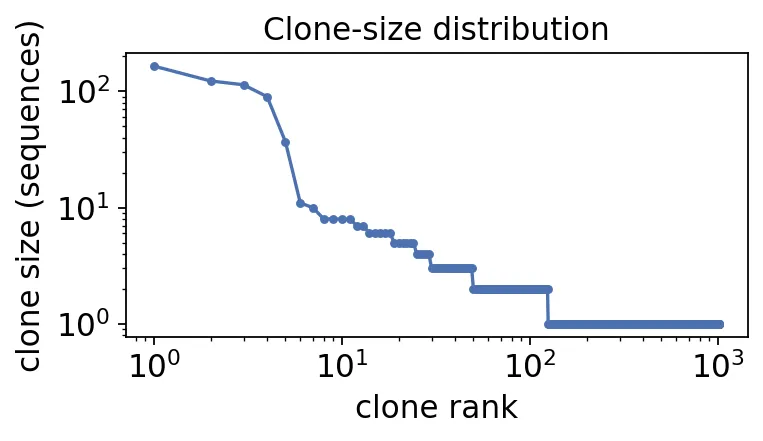 BCR clone size rank curve
BCR clone size rank curve大多数B细胞克隆都是singleton,也就是只采到一条序列。
但真正有意思的是那49个expanded clones,尤其是最大的克隆有165条序列。这种大克隆一般不会凭空出现,通常对应抗原刺激后的扩增。
5. 扩增克隆是不是疫苗反应
如果这些大克隆来自疫苗反应,那么它们应该主要出现在第7天,而不是疫苗接种前。
tp = ov.airr.clone_timepoint_distribution(clones, min_size=8)
print(tp.head(8))
day7_frac = tp.attrs['timepoint_share']['+7d']
print(f"\nday-7 share of the largest clones: {day7_frac:.0%}")
sample_id +7d -1h
clone_id
1010 165 0
1016 123 0
1013 114 0
1011 90 0
1014 37 0
1012 11 0
940 10 0
1015 8 0
day-7 share of the largest clones: 98%
这个结果非常干净。
最大的几个克隆几乎全部来自疫苗后第7天。也就是说,我们前面看到的B细胞克隆扩增,基本就是疫苗反应在IgH repertoire里的痕迹。
6. 克隆丰度和多样性
接下来,我们用rank-abundance曲线看两个时间点的克隆丰度结构。
abund, abund_df = ov.airr.clonal_abundance(
clones,
clone='clone_id',
group='sample_id',
nboot=200,
seed=0,
as_curve_data=True,
)
top = abund_df.sort_values(['sample_id', 'rank']).groupby('sample_id').head(3)
print("top-3 clones per timepoint (rarefied abundance +/- CI):")
print(top[['sample_id', 'rank', 'p', 'lower', 'upper']].round(3).to_string(index=False))
top-3 clones per timepoint (rarefied abundance +/- CI):
sample_id rank p lower upper
+7d 1 0.194 0.166 0.222
+7d 2 0.146 0.123 0.168
+7d 3 0.136 0.114 0.159
-1h 1 0.008 0.002 0.015
-1h 2 0.008 0.001 0.014
-1h 3 0.007 0.001 0.013
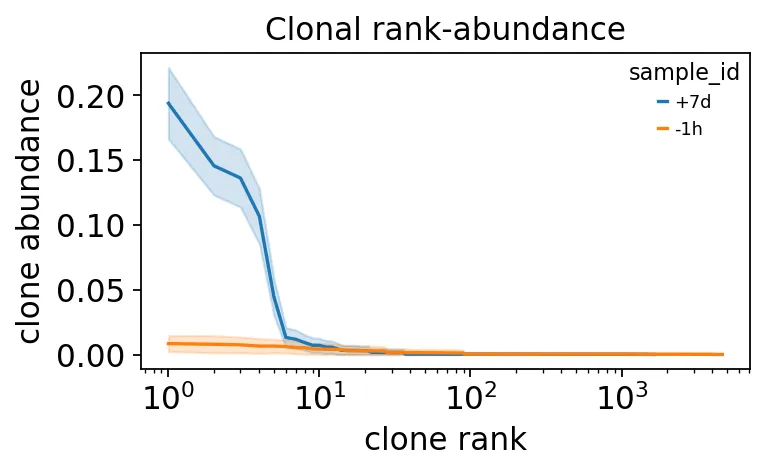 克隆rank-abundance曲线
克隆rank-abundance曲线这张图很直观。
疫苗后第7天的rank-abundance曲线非常陡,说明前几个克隆占据了很大比例。接种前曲线则很平,没有明显的大克隆支配。
我们再用Hill diversity看多样性:
div = ov.airr.hill_diversity(
clones,
group='sample_id',
min_q=0,
max_q=4,
step_q=0.25,
)
dtab = div.diversity
q_show = dtab[dtab['q'].isin([0, 1, 2])]
print("Hill diversity at q = 0 (richness), 1 (Shannon), 2 (Simpson):")
print(q_show[['sample_id', 'q', 'd', 'd_lower', 'd_upper']].round(1).to_string(index=False))
Hill diversity at q = 0 (richness), 1 (Shannon), 2 (Simpson):
sample_id q d d_lower d_upper
+7d 0.0 235.1 211.5 258.6
+7d 1.0 34.9 28.9 40.8
+7d 2.0 10.8 9.6 12.0
-1h 0.0 693.3 671.3 715.4
-1h 1.0 619.8 585.7 653.8
-1h 2.0 501.9 444.8 559.1
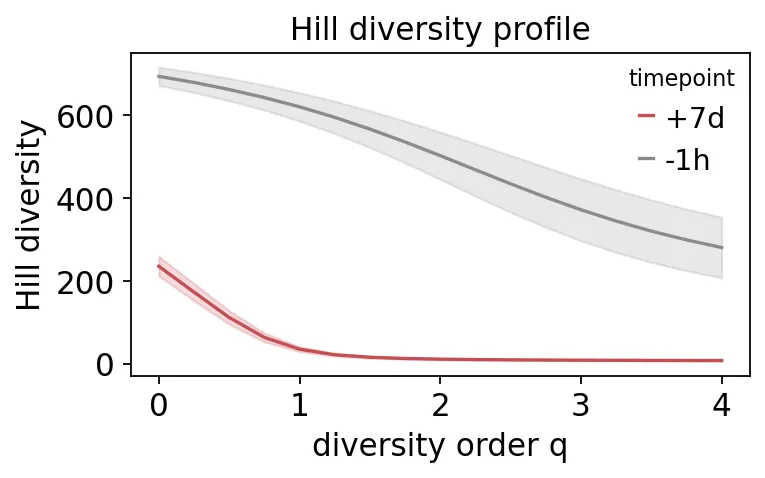 Hill diversity profile
Hill diversity profile第7天的多样性明显下降,尤其是q从0升高后下降得更快。
这说明疫苗后不是所有B细胞都平均扩增。少数抗原相关克隆被拉起来以后,repertoire自然就变得更“偏心”。
7. Germline重建
BCR分析里,germline非常重要。
因为我们要判断一条BCR突变了多少,就必须知道它最初没有突变时的germline序列是什么。
这个Laserson数据已经带好了germline列,所以教程里先检查这些列是否存在:
gl_cols = ['germline_alignment', 'germline_alignment_d_mask']
present = [c for c in gl_cols if c in clones.columns]
print(f"germline columns present: {present}")
print(f"germline length: {clones['germline_alignment'].dropna().str.len().iloc[0]} bp")
print(f"D-region positions masked with 'N': {(clones['germline_alignment_d_mask'].iloc[0] == 'N').sum()}")
print(f"non-null germlines: {clones['germline_alignment'].notna().sum()} / {len(clones)} sequences")
germline columns present: ['germline_alignment', 'germline_alignment_d_mask']
germline length: 433 bp
D-region positions masked with 'N': 79
non-null germlines: 1780 / 1780 sequences
如果你要从头跑自己的数据,一般需要提供完整IMGT V/D/J参考,然后运行:
import pydowser
references = pydowser.readIMGT('/path/to/IMGT/human/vdj')
clones = ov.airr.reconstruct_germlines(clones, references=references)
后面的SHM、BASELINe和谱系树,都依赖“观测序列 vs germline序列”的比较。如果germline错了,后面的突变和选择都会跟着错。
8. 体细胞高频突变
接下来计算SHM。
mut = ov.airr.mutation_analysis(
clones,
frequency=True,
combine=True,
)
mut = mut.rename(columns={'mu_freq': 'shm_freq'})
print(f"mean SHM frequency: {mut['shm_freq'].mean():.3f} ({mut['shm_freq'].mean()*100:.1f}% of V-region bases mutated)")
print(f"range: {mut['shm_freq'].min():.3f} - {mut['shm_freq'].max():.3f}")
mean SHM frequency: 0.041 (4.1% of V-region bases mutated)
range: 0.000 - 0.218
然后按时间点和isotype拆开看:
by_time = ov.airr.summarize_by_group(
mut['shm_freq'],
group=mut['sample_id'],
agg='mean',
)['shm_freq']
by_iso = ov.airr.summarize_by_group(
mut['shm_freq'],
group=mut['c_call'],
agg='mean',
)['shm_freq']
mean SHM by timepoint:
sample_id
+7d 6.6 %
-1h 1.9 %
mean SHM by isotype:
c_call
IGHA 7.7 %
IGHD 0.5 %
IGHG 7.8 %
IGHM 0.9 %
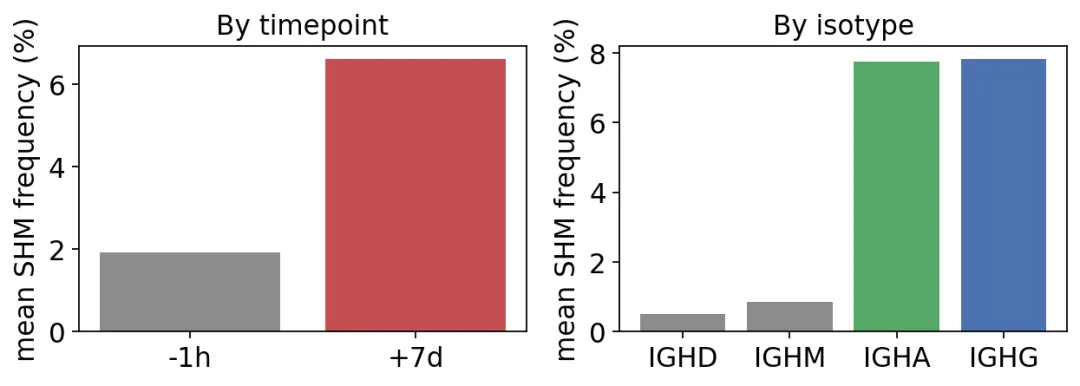 SHM按时间点和isotype分布
SHM按时间点和isotype分布疫苗后第7天的SHM从1.9%升到6.6%。同时,IgG和IgA的SHM大约是7.8%和7.7%,IgM和IgD则只有0.9%和0.5%。
这说明第7天的BCR不只是数量上扩增了,它们也确实带着更多生发中心反应后的突变。
9. 突变落在V基因的哪里
只看整体SHM频率还不够。
抗体V基因里有FWR和CDR区域。FWR更像结构支架,CDR更靠近抗原接触面。
如果亲和力成熟真的发生了,我们会希望看到CDR区域积累更多突变,而FWR区域相对更保守。
mut_reg = ov.airr.mutation_analysis(
clones,
frequency=True,
combine=False,
region='v',
)
regions = ['fwr1', 'cdr1', 'fwr2', 'cdr2', 'fwr3']
print("mean SHM frequency by IMGT V-region:")
for r in regions:
print(f" {r.upper():5s}: {mut_reg[f'{r}_mu_freq'].mean()*100:.2f} %")
mean SHM frequency by IMGT V-region:
FWR1 : 2.65 %
CDR1 : 5.05 %
FWR2 : 2.95 %
CDR2 : 7.99 %
FWR3 : 4.24 %
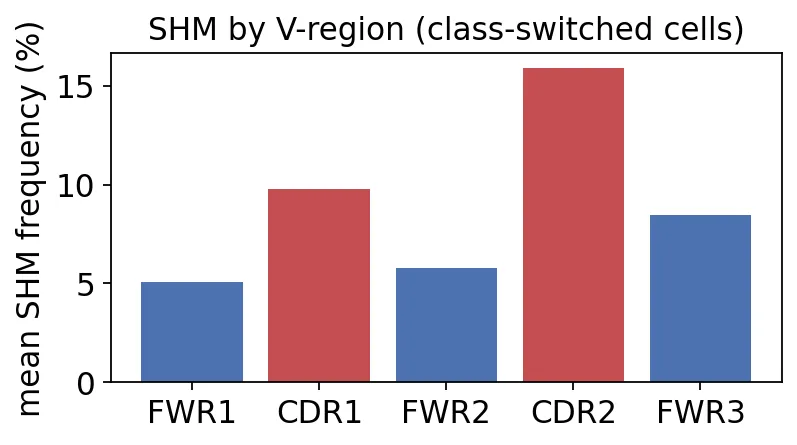 V基因不同区域SHM频率
V基因不同区域SHM频率可以看到,CDR1和CDR2的突变频率更高,尤其是CDR2接近8%。
这符合亲和力成熟的直觉:CDR区域更可能直接影响抗原结合,所以突变更容易被保留下来;FWR区域要维持抗体结构,很多replacement mutation会被淘汰。
10. BASELINe抗原选择分析
SHM告诉我们“突变发生了多少”,BASELINe进一步问的是:这些突变有没有被选择?
它的思路大概是这样:
- silent mutation通常不改变氨基酸,可以用来估计中性突变背景。
- replacement mutation会改变氨基酸,可能影响抗原结合或抗体结构。
- 如果某一区域的replacement mutation比中性预期更多,说明可能存在正选择。
- 如果某一区域的replacement mutation比中性预期更少,说明可能存在负选择或纯化选择。
这里我们只分析expanded clones,因为只有扩增并积累足够突变的克隆,才适合做选择分析。
expanded = clones[clones['clone_id'].isin(sizes[sizes >= 3].index)].copy()
print(
f"expanded-clone sequences: {len(expanded)} "
f"in {expanded['clone_id'].nunique()} clones"
)
selection = ov.airr.baseline_selection(expanded, region='v')
selection.drop_duplicates('region')
expanded-clone sequences: 736 in 49 clones
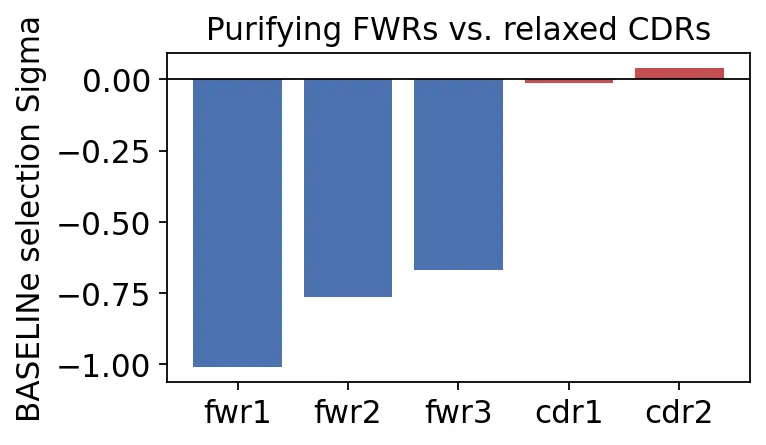 BASELINe selection sigma
BASELINe selection sigmaFWR1、FWR2、FWR3都是明显负Sigma,说明framework区域存在纯化选择。也就是说,破坏抗体结构的replacement mutation更容易被清除。
CDR1和CDR2接近0,没有明显负选择。这里不是说CDR没有用。更合理的理解是,CDR不像FWR那样受到强结构约束,它更能容忍改变,也更可能积累影响抗原结合的突变。
如果只看SHM频率,我们只能说“突变更多”。加上BASELINe后,我们能进一步说:突变和选择的空间分布符合抗原驱动的亲和力成熟。
11. B细胞谱系树
对BCR来说,谱系树是一个很自然的分析。
同一个B细胞克隆内部,不同序列之间的突变关系可以组成一棵树。根节点对应推断出的未突变共同祖先,分支代表SHM积累,叶子是实际观测到的序列。
pick = sizes[(sizes >= 8) & (sizes <= 37)].index[:5]
tree_in = ov.airr.collapse_germlines(
clones[clones['clone_id'].isin(pick)].copy()
)
print(f"building lineage trees for {len(pick)} clones")
building lineage trees for 5 clones
trees = ov.airr.lineage_trees(tree_in, build='pratchet')
summary = pd.DataFrame({
'clone_id': trees['clone_id'],
'n_tips': [t.n_tip for t in trees['tree']],
'tree_length': [t.length for t in trees['tree']],
})
summary
clone_id n_tips tree_length
1014 19 0.182
940 10 0.168
107 7 0.102
1015 6 0.078
1012 5 0.081
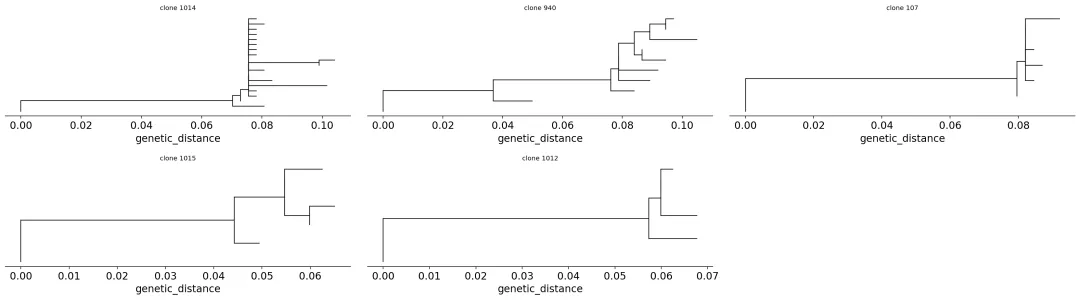 B细胞谱系树
B细胞谱系树在这张图上,我们把亲和力成熟过程画了出来。
如果一个克隆只有很多完全相同的序列,那它只是扩增。现在我们看到的是分叉的谱系树,说明这个克隆在扩增过程中还继续积累了突变,并产生了多个相近但不同的亚分支。
这就是BCR分析比TCR分析更像“进化分析”的地方。
12. IGHV基因型
BCR的突变分析还有一个容易被忽略的问题:每个人的IGHV germline allele不完全一样。
如果你用错了参考等位基因,本来属于这个人的germline差异就会被误判成SHM。
因此,在正式流程里,应该先做TIgGER genotype inference,再做克隆推断和后续SHM分析。这个教程为了叙事顺序,把基因型部分放在后面,但真实项目里顺序应该提前。
import pytigger
geno_in = ov.airr.normalize_gene_calls(clones, cols=('v_call', 'j_call'))
germline_ighv = pytigger.load_sample_germline_ighv()
print(f"IMGT germline IGHV reference: {len(germline_ighv)} alleles")
IMGT germline IGHV reference: 344 alleles
genotype = ov.airr.infer_genotype(
geno_in,
germline_db=germline_ighv,
method='frequency',
)
print(f"IGHV genes in this subject's genotype: {genotype.shape[0]}")
print(f"heterozygous genes (>1 allele): {(genotype['alleles'].str.contains(',')).sum()}")
genotype.head()
IGHV genes in this subject's genotype: 40
heterozygous genes (>1 allele): 9
然后检测是否存在候选novel allele:
novel = ov.airr.find_novel_alleles(geno_in, germline_ighv)
n_novel = int(novel['polymorphism_call'].notna().sum())
print(f"candidate novel IGHV alleles: {n_novel}")
print(f"genes screened: {novel.shape[0]}")
candidate novel IGHV alleles: 0
genes screened: 1
这个示例数据里没有发现novel allele。对教程来说,这个结果没有太多故事性,但它提醒我们一件事:BCR SHM分析不能随便拿一个germline reference就跑,基因型最好先确认。
13. Isotype switching和CDR3性质
最后看isotype组成。
iso_frac = ov.airr.isotype_composition(clones, group='sample_id')
print("isotype fraction by timepoint:")
print(iso_frac.round(3).to_string())
switch = iso_frac[['IGHA', 'IGHG']].sum(axis=1)
print(
f"class-switched (IgG+IgA) fraction: "
f"-1h {switch['-1h']:.0%} -> +7d {switch['+7d']:.0%}"
)
isotype fraction by timepoint:
c_call IGHA IGHD IGHG IGHM
sample_id
+7d 0.248 0.057 0.530 0.165
-1h 0.099 0.207 0.116 0.577
class-switched (IgG+IgA) fraction: -1h 22% -> +7d 78%
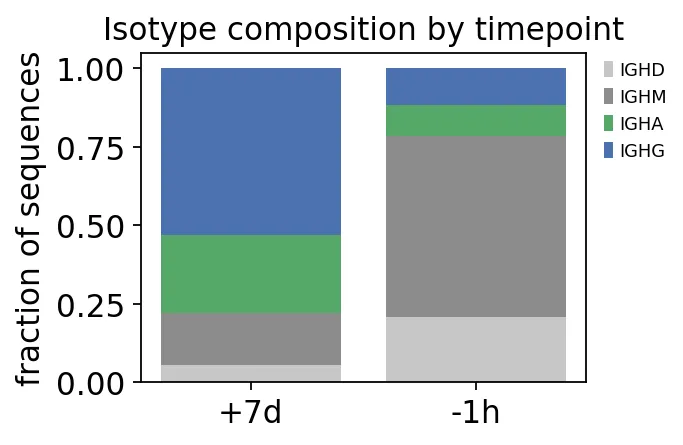 isotype composition
isotype composition这个结果和前面的SHM分析能对上。
接种前以IgM为主,IgG+IgA占比只有22%;第7天IgG+IgA上升到78%。也就是说,第7天的repertoire确实被class-switched B细胞占据。
然后我们可以看CDR3氨基酸性质:
aap = ov.airr.aa_properties(clones, seq='junction', nt=True)
prop_cols = [
'junction_aa_length',
'junction_aa_gravy',
'junction_aa_charge',
'junction_aa_polarity',
]
print("mean CDR3 amino-acid properties by isotype:")
print(aap.groupby('c_call')[prop_cols].mean().round(3).to_string())
mean CDR3 amino-acid properties by isotype:
junction_aa_length junction_aa_gravy junction_aa_charge junction_aa_polarity
c_call
IGHD 23.314 -0.363 -0.769 8.011
IGHM 22.738 -0.401 -0.712 8.104
IGHA 20.228 -0.295 -0.399 8.317
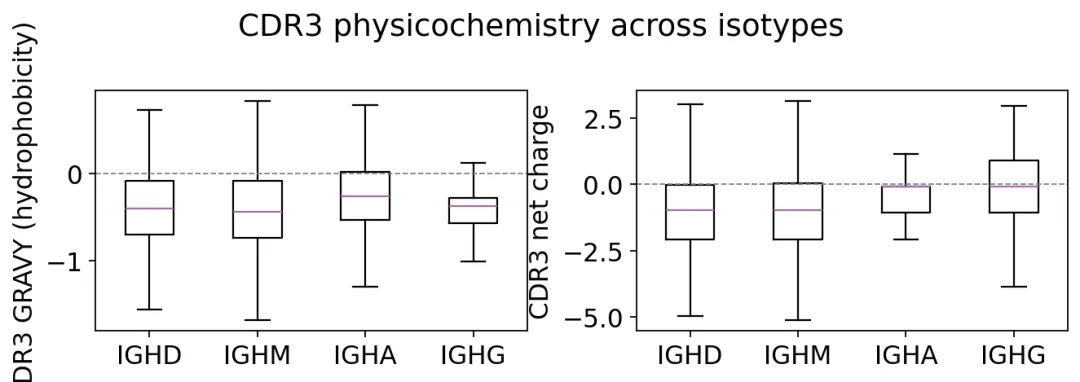 CDR3理化性质
CDR3理化性质CDR3性质图告诉我们不同isotype对应的抗原结合环在长度、疏水性、电荷上有没有偏移。
14. 总结
在本期教程中,我们用Laserson 2014流感疫苗IgH数据,重建了一次B细胞亲和力成熟过程:
- 原始数据包含1999条IgH序列,保留productive rearrangements后剩下1780条。
- 数据包含疫苗接种前1小时和接种后第7天两个时间点。
- 一共推断出1018个B细胞克隆,其中894个是singleton,49个是expanded clones,最大克隆包含165条序列。
- 最大的一批克隆98%来自第7天,说明它们基本对应疫苗后的克隆扩增。
- 第7天rank-abundance曲线明显更陡,Hill diversity也明显下降,说明少数克隆占据了repertoire。
- 平均SHM从接种前1.9%升到第7天6.6%,IgG和IgA的SHM也远高于IgM和IgD。
- CDR1/CDR2区域的突变频率高于FWR区域,尤其CDR2达到7.99%。
- BASELINe显示FWR1、FWR2、FWR3存在明显负选择,说明framework区域的replacement mutation受到清除。
- IgG+IgA比例从接种前22%升到第7天78%,和SHM升高、克隆扩增一起构成了典型疫苗反应。
实际上我觉得BCR分析最有意思的地方,是它可以把免疫反应拆成一条时间线。
接种前,repertoire比较分散,IgM/IgD占比高,SHM低。接种后第7天,几个大克隆被拉起来,IgG/IgA比例升高,SHM增加,FWR受到纯化选择,谱系树也开始分叉。
这些结果合在一起,就是一次B细胞亲和力成熟的分子记录。
如果你做的是疫苗、感染、自身免疫病或者抗体发现项目,BCR repertoire分析一般都会绕不开这些内容。克隆扩增只是第一步,真正能把故事讲完整的,还是SHM、selection、lineage tree和isotype switching这些B细胞特有的分析。
15. 交流群
如果你也很好奇我们omicverse生态的最新进展,想第一时间体验到新功能,欢迎加入我们的交流群~

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
