为什么要进行聚类?
在上一节中,我们已经完成了 PCA、邻居图和 UMAP。此时每个细胞已经不再只是一个高维基因表达向量,而是在 PCA 空间和邻居图中有了相对清晰的位置关系。
聚类(clustering)的目的,是在这些细胞之间的相似性关系中,找到表达模式相近的一组细胞。简单理解就是:
细胞邻居图 → 细胞群
在单细胞 RNA-seq 分析中,聚类通常用于帮助我们发现数据中的主要细胞群、细胞状态或潜在亚群。它本身不是最终的细胞类型注释,而是为后续 marker gene 分析和细胞类型注释提供一个分组基础。
需要注意的是,聚类结果不等同于真实细胞类型。一个真实细胞类型可能会因为状态差异、细胞周期、激活状态或技术因素被拆成多个 cluster;多个相近细胞类型也可能在较低分辨率下被合并到同一个 cluster。因此,聚类结果需要结合 marker gene、样本信息、QC 指标和生物学背景进行解释。
聚类的基本思想
在 Scanpy 的常规流程中,聚类不是直接在原始表达矩阵上进行,也不是在 UMAP 二维坐标上进行,而是在邻居图上进行。
上一节降维中的核心步骤是:
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=30,
random_state=0
)
这一步会在 PCA 空间中寻找每个细胞的近邻,并构建细胞之间的 KNN graph。可以把这个图理解为一个由细胞构成的网络:
Leiden 聚类算法会在这个邻居图上寻找连接更紧密的细胞社区,也就是我们后续看到的 cluster。
因此需要特别区分:
PCA 空间 → 构建邻居图 → Leiden 聚类
PCA 空间 → 构建邻居图 → UMAP 可视化
UMAP 和 Leiden 都依赖邻居图,但二者用途不同。UMAP 主要用于可视化,Leiden 用于产生聚类标签。不要直接根据 UMAP 二维图上的距离或形状来定义 cluster。
Leiden 和 Louvain
Leiden 和 Louvain 都是图聚类算法,常用于在单细胞邻居图上发现细胞群。Louvain 是较早使用的方法,Leiden 是对 Louvain 的改进。
目前单细胞分析中通常优先使用 Leiden。相比 Louvain,Leiden 更稳定,也能减少一个 cluster 内部并不连通的问题。因此在新的 Scanpy 流程中,建议优先使用:
sc.tl.leiden()
Louvain 可以作为历史方法或对比方法了解,但一般不作为首选。
读取降维后的数据
import os
import pandas as pd
import scanpy as sc
adata = sc.read_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/05_pbmc_10k_dimensionality_reduction.h5ad")
adata
这里读入的是上一节保存的降维结果。这个对象应该已经包含 PCA、邻居图和 UMAP 结果。
可以先检查对象中是否已经有这些信息:
print(adata.obsm.keys())
print(adata.obsp.keys())
print(adata.uns.keys())
通常应该能看到:
adata.obsm["X_pca"]:PCA 坐标。adata.obsm["X_umap"]:UMAP 坐标。adata.obsp["distances"]:邻居距离矩阵。adata.obsp["connectivities"]:邻居连接矩阵。adata.uns["neighbors"]:邻居图参数。
如果当前对象没有邻居图,可以重新运行:
n_pcs = 30
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=n_pcs,
random_state=0
)
如果上一节已经保存了邻居图,这一步可以不用重复运行。
运行 Leiden 聚类
先从一个常用分辨率开始,例如 resolution=0.5:
sc.tl.leiden(
adata,
resolution=0.5,
key_added="leiden_res0_5",
flavor="igraph",
n_iterations=2,
random_state=0
)
这里几个参数的含义是:
resolution=0.5:控制聚类颗粒度。值越高,通常得到的 cluster 越多;值越低,cluster 越少。key_added="leiden_res0_5":将聚类结果保存到 adata.obs["leiden_res0_5"] 中,避免覆盖其他分辨率的结果。flavor="igraph":使用 igraph 后端运行 Leiden。n_iterations=2:运行 2 次优化迭代,是常用且效率较高的设置。random_state=0:固定随机种子,方便复现结果。
执行完成后,聚类标签会被写入 adata.obs:
adata.obs[["leiden_res0_5"]].head()
查看每个 cluster 的细胞数:
adata.obs["leiden_res0_5"].value_counts().sort_index()
也可以整理成表格:
cluster_key = "leiden_res0_5"
cluster_counts = adata.obs[cluster_key].value_counts().sort_index().to_frame("n_cells")
cluster_counts["pct_cells"] = cluster_counts["n_cells"] / adata.n_obs * 100
cluster_counts
如果某个 cluster 细胞数非常少,需要特别谨慎。它可能是真实稀有细胞群,也可能是低质量细胞、doublet、批次效应或聚类分辨率过高造成的小群。
在 UMAP 上查看聚类结果
聚类结果通常会画在 UMAP 上进行观察:
sc.pl.umap(
adata,
color="leiden_res0_5",
legend_loc="on data"
)
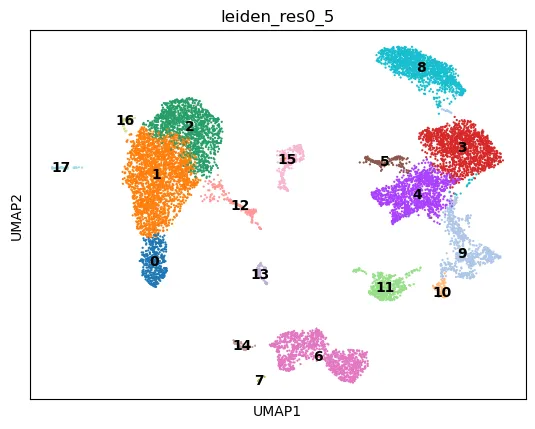
这里需要再次强调:Leiden 聚类不是在 UMAP 坐标上运行的。UMAP 只是把聚类标签显示出来,方便我们观察不同 cluster 在二维空间中的分布。
如果想让图例放在图外,可以使用:
sc.pl.umap(
adata,
color="leiden_res0_5",
legend_loc="right margin"
)
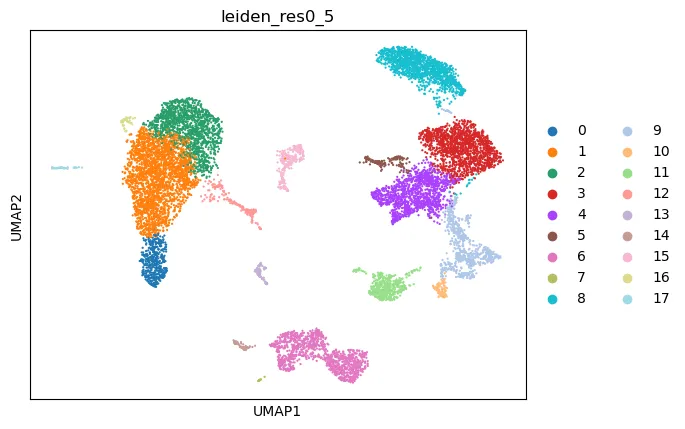
尝试不同分辨率
单细胞聚类中通常不会只看一个 resolution。因为不同分辨率会得到不同颗粒度的细胞群。
低分辨率更适合观察粗略的主要细胞类型;高分辨率更适合发现细胞状态或亚群,但也更容易把噪音拆成小 cluster。
可以一次运行多个分辨率,并把结果保存到不同列中:
resolution_settings = {
"leiden_res0_25": 0.25,
"leiden_res0_5": 0.5,
"leiden_res0_8": 0.8,
"leiden_res1": 1.0,
"leiden_res1_5": 1.5,
}
for key, res in resolution_settings.items():
sc.tl.leiden(
adata,
resolution=res,
key_added=key,
flavor="igraph",
n_iterations=2,
random_state=0
)
print(key, adata.obs[key].nunique())
然后把不同分辨率画在同一张 UMAP 上进行比较:
sc.pl.umap(
adata,
color=["leiden_res0_25", "leiden_res0_5", "leiden_res0_8", "leiden_res1"],
legend_loc="on data",
ncols=2
)
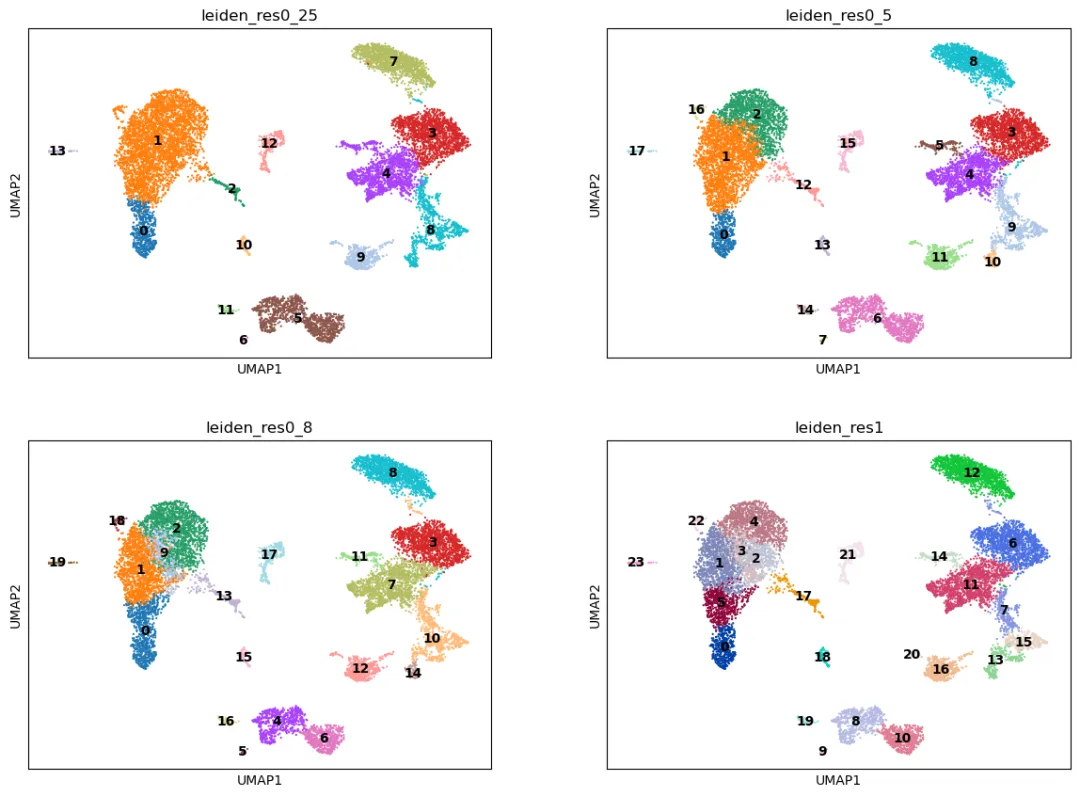
判断分辨率时,不应该只看 cluster 数量,而应该结合后面的 marker gene 和 QC 信息:
- 如果一个 cluster 中明显混合了两类互斥 marker,可能说明分辨率太低,需要拆开。
- 如果多个小 cluster 的 marker gene 几乎完全一样,可能说明分辨率太高,可以合并解释。
- 如果某个小 cluster 主要由高线粒体比例、极端 UMI 或某个批次组成,应优先考虑技术因素。
对于 PBMC 这类细胞类型相对清楚的数据,可以先从 resolution=0.5 或 resolution=0.8 开始。最终使用哪个分辨率,应该在 marker gene 分析和细胞类型注释之后再确定。
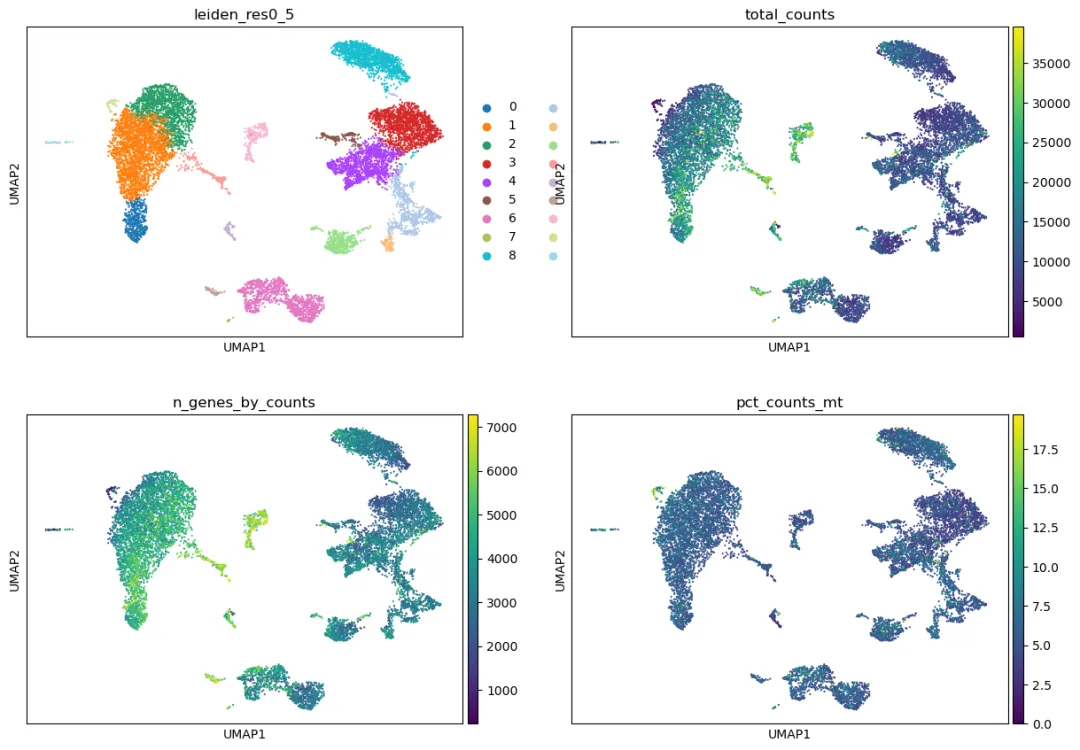
检查聚类结果是否被 QC 指标驱动
聚类完成后,第一步不是马上给 cluster 命名,而是检查 cluster 是否主要由技术因素驱动。
先用 QC 指标给 UMAP 上色:
qc_cols = [col for col in ["total_counts", "n_genes_by_counts", "pct_counts_mt"] if col in adata.obs.columns]
if len(qc_cols) > 0:
sc.pl.umap(
adata,
color=["leiden_res0_5"] + qc_cols,
ncols=2
)
还可以按 cluster 计算 QC 指标的中位数:
cluster_key = "leiden_res0_5"
adata.obs.groupby(cluster_key, observed=True)[
["total_counts", "n_genes_by_counts", "pct_counts_mt"]
].median().sort_values("pct_counts_mt", ascending=False)
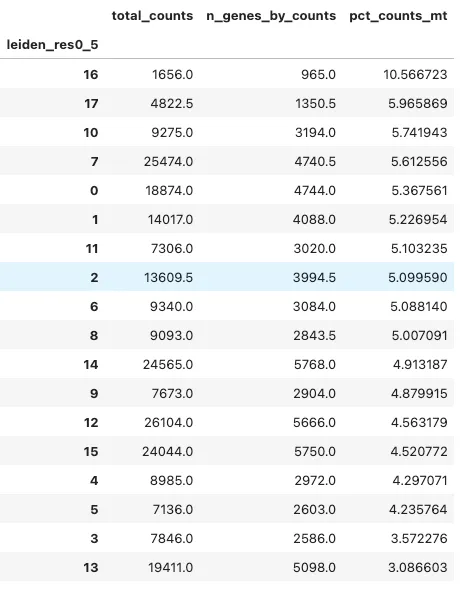
如果某个 cluster 的 pct_counts_mt 明显偏高,或者 total_counts、n_genes_by_counts 极端异常,需要考虑它是否是低质量细胞、doublet 或残留技术噪音。
如果对象中有样本或批次信息,也应该检查 cluster 是否被样本或批次主导:
sample_cols = [col for col in ["sample", "batch"] if col in adata.obs.columns]
for col in sample_cols:
print(col)
print(pd.crosstab(adata.obs[cluster_key], adata.obs[col], normalize="index"))
如果某个 cluster 几乎只来自一个样本或一个批次,需要结合实验设计判断它是真实生物学差异,还是批次效应。
使用 marker gene 初步检查聚类
对于 PBMC 数据,可以先用一些经典 marker gene 粗略检查聚类是否合理。
marker_genes = ["MS4A1", "CD3D", "CD8A", "LYZ", "NKG7", "PPBP"]
if adata.raw isnotNone:
marker_genes = [gene for gene in marker_genes if gene in adata.raw.var_names]
if len(marker_genes) > 0:
sc.pl.umap(
adata,
color=["leiden_res0_5"] + marker_genes,
use_raw=True,
ncols=3
)
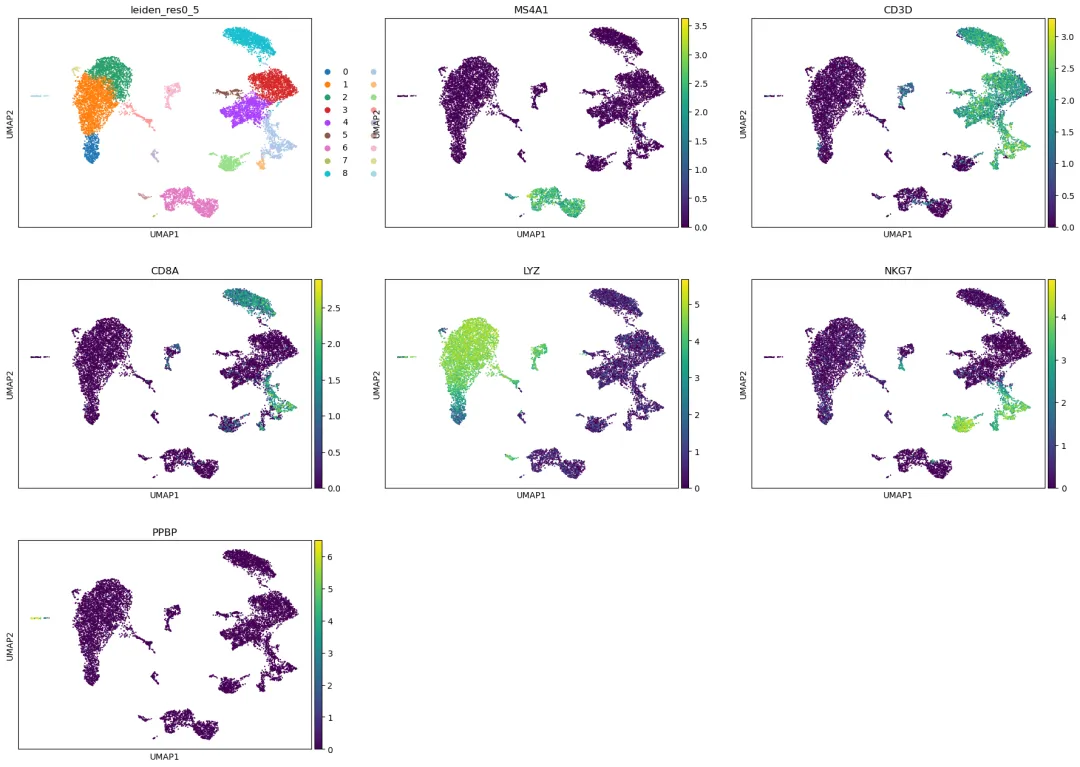
这里使用 use_raw=True,是为了展示 Log(CP10k+1) 后的表达值,而不是 scaling 后的 z-score。
常见 PBMC marker 可以这样初步理解,但不同的组织样本可能有不同的 marker:
NKG7:NK cells 或 cytotoxic T cells。
这一步只是初步检查,不等同于正式注释。正式注释通常需要在每个 cluster 中寻找 marker gene,并结合已有细胞类型知识进行判断。
如何选择最终聚类结果?
在实际分析中,常常会保存多个分辨率的 Leiden 结果,但最终注释时需要选择一个主要使用的聚类标签。
可以先选一个中等分辨率作为起点,例如:
cluster_key = "leiden_res0_5"
如果后续 marker gene 分析发现某些 cluster 内部仍然混有明显不同的细胞类型,可以尝试更高分辨率,例如:
cluster_key = "leiden_res0_8"
如果高分辨率拆出了很多 marker gene 几乎相同的小 cluster,则可以回到较低分辨率,或者在注释时把相近 cluster 合并解释。
需要注意的是,resolution 不是一个越高越好的参数。它控制的是算法划分细胞社区的细度,而不是细胞类型的真实数量。
亚群再聚类(可选)
有时我们会对某一个大类细胞进行更细致的分析。例如,在完成全局聚类和初步注释后,只取出 T cells,再重新进行邻居图、UMAP 和 Leiden 聚类,以寻找 T cell 内部的亚群或状态。
示例代码如下:
cluster_key = "leiden_res0_5"
target_cluster = "3"
adata_sub = adata[adata.obs[cluster_key] == target_cluster].copy()
adata_sub
对子集重新构建邻居图并聚类:
sc.pp.neighbors(
adata_sub,
n_neighbors=15,
n_pcs=30,
random_state=0
)
sc.tl.umap(
adata_sub,
min_dist=0.3,
spread=1.0,
random_state=0
)
sc.tl.leiden(
adata_sub,
resolution=0.5,
key_added="leiden_sub",
flavor="igraph",
n_iterations=2,
random_state=0
)
查看子集聚类结果:
sc.pl.umap(
adata_sub,
color="leiden_sub",
legend_loc="on data"
)
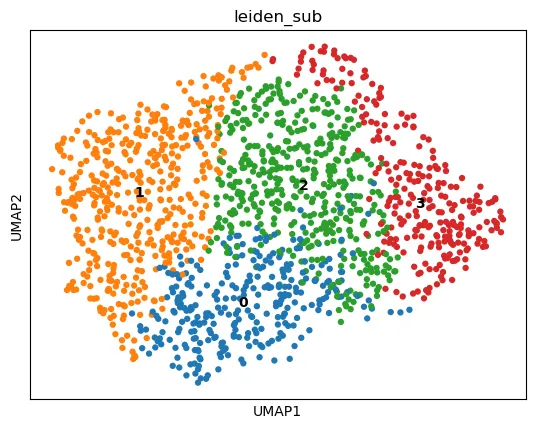
亚群再聚类适合在已经有明确细胞大类之后使用。不要在还没有完成基础 QC、批次检查和全局聚类解释之前,过早进行过细的亚群拆分。
保存
保存完成聚类后的对象:
adata.write_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/06_pbmc_10k_clustered.h5ad")
检查文件是否保存成功:
os.path.exists("/home/data/t090639/project/pbmc_10k/data/processed/06_pbmc_10k_clustered.h5ad")
保存后的对象中应至少包含:
adata.obs["leiden_res0_25"]:低分辨率 Leiden 聚类结果。adata.obs["leiden_res0_5"]:中等分辨率 Leiden 聚类结果。adata.obs["leiden_res0_8"]:较高分辨率 Leiden 聚类结果。adata.obs["leiden_res1"]:高分辨率 Leiden 聚类结果。adata.obsm["X_umap"]:UMAP 坐标。adata.obsp["distances"]:邻居距离矩阵。adata.obsp["connectivities"]:邻居连接矩阵。adata.uns["neighbors"]:邻居图参数。
下一章可以基于这里保存的聚类结果继续进行 marker gene 筛选和细胞类型注释。

