python版单细胞注释:基于已知marker基因注释
- 2026-07-04 04:45:03
前面看到一个学妹在学习python,她的一篇推文中提到了一个python代码的学习资源,今天来看看里面的细胞注释部分。
https://www.sc-best-practices.org/preamble.html
So what is a cell type?
什么是细胞类型?
生物学家使用"细胞类型"这一术语指代在数据集中具有稳定性、可通过特定标记物(如蛋白质或基因转录本)表达进行识别,且常与特定功能相关联的细胞表型。例如,浆细胞B细胞是一种能分泌抗体对抗病原体的白细胞类型,可通过特定标记物进行识别。了解样本中存在的细胞类型对理解数据至关重要。比如,发现肿瘤中存在特定免疫细胞类型,或骨髓样本中出现异常造血干细胞,可能为您研究的疾病提供宝贵见解。
专业词汇:“subtypes” or “cell states” ,“cell identity”,三者往往混用有区别又有联系,看下面这两个文献对其的讨论与定义:
Revealing the vectors of cellular identity with single-cell genomics. Nature Biotechnology
Hongkui Zeng. What is a cell type and how to define it? Cell
那么,在单细胞数据中如何进行细胞注释?注释方法多种多样,下文将概述不同策略。由于我们处理的是转录组数据,所有方法最终都基于特定基因或基因集的表达,以及细胞间的整体转录组相似性。
图来源:https://www.worksheetsplanet.com/what-is-a-cell/
1 环境配置
过滤掉不影响代码运行的弃用警告和性能警告:
import warningswarnings.filterwarnings("ignore", category=DeprecationWarning)from numba.core.errors import NumbaDeprecationWarningwarnings.simplefilter("ignore", category=NumbaDeprecationWarning)导入模块:
我都装在了conda小环境 sc中,这里有个自动注释的模块很难配置(scarches),但是本期没有安装上不会影响下面的内容。
import urllib.requestfrom pathlib import Pathimport celltypistimport matplotlib.pyplot as pltimport numpy as npimport pandas as pdimport scanpy as scimport scarches as scaimport seaborn as snsfrom celltypist import modelsfrom scipy.sparse import csr_matrix2 示例数据
数据来自这里,https://openreview.net/forum?id=gN35BGa1Rt,该数据集捕获了来自12位健康人类供者的骨髓单核细胞单细胞多组学数据。
下载地址:
https://figshare.com/ndownloader/files/41436666
3 细胞注释
两种路径:
从已知细胞类型标记基因表出发 → 匹配至聚类群组 从聚类的高表达基因出发 → 关联至已知细胞类型
3.1 从标记基因到聚类注释
首先列出一组基于文献的骨髓细胞类型标记基因:这些标记来自以往研究特定细胞类型和亚型、并报道了相应标记基因的论文。需要注意的是,蛋白质水平的标记(例如用于流式细胞术的标记)有时在转录组数据中效果不佳,因此使用基于RNA研究的论文中的标记通常更有效。
理想情况下,标记基因集应在多个数据集中得到验证。
此外,与领域专家合作通常很有帮助:作为生物信息学家,尝试与更深入了解组织特性、生物学背景、预期细胞类型和标记基因的生物学家合作。
marker_genes = {"CD14+ Mono": ["FCN1", "CD14"],"CD16+ Mono": ["TCF7L2", "FCGR3A", "LYN"],"ID2-hi myeloid prog": ["CD14", "ID2", "VCAN", "S100A9", "CLEC12A", "KLF4", "PLAUR"],"cDC1": ["CLEC9A", "CADM1"],"cDC2": ["CST3", "COTL1", "LYZ", "DMXL2", "CLEC10A", "FCER1A"], # Note: DMXL2 should be negative"Normoblast": ["SLC4A1", "SLC25A37", "HBB", "HBA2", "HBA1", "TFRC"],"Erythroblast": ["MKI67", "HBA1", "HBB"],"Proerythroblast": ["CDK6", "SYNGR1", "HBM", "GYPA"], # Note HBM and GYPA are negative markers"NK": ["GNLY", "NKG7", "CD247", "GRIK4", "FCER1G", "TYROBP", "KLRG1", "FCGR3A"],"ILC": ["ID2", "PLCG2", "GNLY", "SYNE1"],"Lymph prog": ["VPREB1", "MME", "EBF1", "SSBP2", "BACH2", "CD79B", "IGHM", "PAX5", "PRKCE", "DNTT", "IGLL1"],"Naive CD20+ B": ["MS4A1", "IL4R", "IGHD", "FCRL1", "IGHM"],"B1 B": ["MS4A1", "SSPN", "ITGB1", "EPHA4", "COL4A4", "PRDM1", "IRF4", "CD38", "XBP1", "PAX5", "BCL11A", "BLK", "IGHD", "IGHM", "ZNF215"], # Note IGHD and IGHM are negative markers"Transitional B": ["MME", "CD38", "CD24", "ACSM3", "MSI2"],"Plasma cells": ["MZB1", "HSP90B1", "FNDC3B", "PRDM1", "IGKC", "JCHAIN"],"Plasmablast": ["XBP1", "RF4", "PRDM1", "PAX5"], # Note PAX5 is a negative marker"CD4+ T activated": ["CD4", "IL7R", "TRBC2", "ITGB1"],"CD4+ T naive": ["CD4", "IL7R", "TRBC2", "CCR7"],"CD8+ T": ["CD8A", "CD8B", "GZMK", "GZMA", "CCL5", "GZMB", "GZMH", "GZMA"],"T activation": ["CD69", "CD38"], # CD69 much better marker!"T naive": ["LEF1", "CCR7", "TCF7"],"pDC": ["GZMB", "IL3RA", "COBLL1", "TCF4"],"G/M prog": ["MPO", "BCL2", "KCNQ5", "CSF3R"],"HSC": ["NRIP1", "MECOM", "PROM1", "NKAIN2", "CD34"],"MK/E prog": ["ZNF385D", "ITGA2B", "RYR3", "PLCB1"], # Note PLCB1 is a negative marker}筛选出仅在本数据中检测到的标记基因:仅保留在adata对象中能找到的基因作为该细胞类型的标记。
marker_genes_in_data = {}for ct, markers in marker_genes.items(): markers_found = []for marker in markers:if marker in adata.var.index: markers_found.append(marker) marker_genes_in_data[ct] = markers_foundUMAP图展示marker基因的表达
先使用scran对数据进行标准化处理,然后降维:
adata.var["highly_variable"] = adata.var["highly_deviant"]sc.tl.pca(adata, n_comps=50, use_highly_variable=True)sc.pp.neighbors(adata)sc.tl.umap(adata)如展示 展示B细胞/浆细胞亚型的marker基因:
B_plasma_cts = ["Naive CD20+ B","B1 B","Transitional B","Plasma cells","Plasmablast",]# 绘图for ct in B_plasma_cts: print(f"{ct.upper()}:") # print cell subtype name sc.pl.umap( adata, color=marker_genes_in_data[ct], vmin=0, vmax="p99", sort_order=False, frameon=False, cmap="Reds", ) print("\n\n\n") # print white space for legibility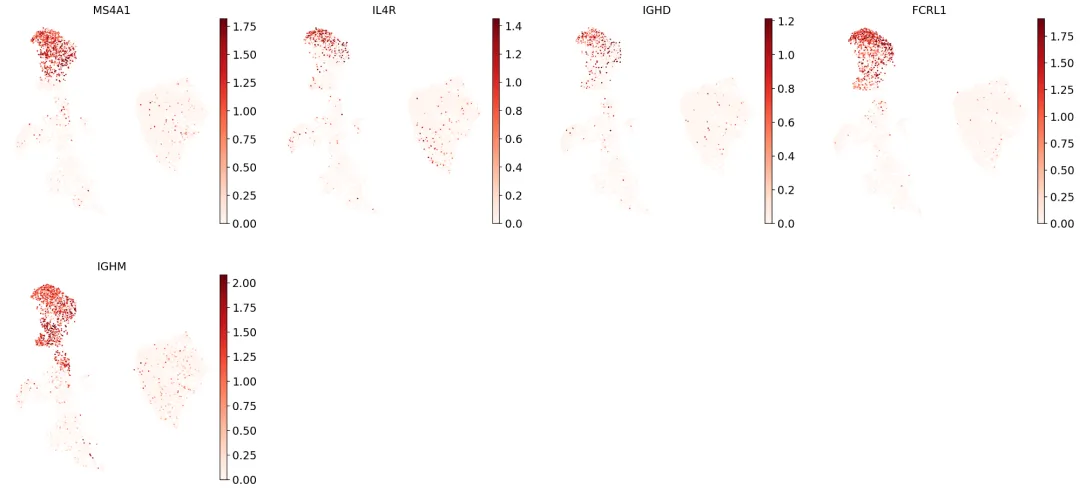
上面共画了5个B细胞亚群,这里放其中一组。从上面(还没有做聚类)可以看出:
标记基因的表达往往较为稀疏,即通常仅在细胞类型的一部分细胞中检测到标记基因表达。这是由于scRNA-seq数据的本质决定的:我们仅对细胞中总RNA分子的一个小子集进行测序,由于这种抽样特性,有时即便基因在细胞中表达,我们也可能无法捕获该基因的转录本。
scRNA-seq细胞注释应基于聚类群体的整体标记基因表达模式,而非依赖单个细胞或基因的表达阈值,以克服数据稀疏性和技术噪声的影响。
此外,其他的色系可选:
https://matplotlib.org/stable/tutorials/colors/colormaps.html
聚类
# 测试多个分辨率并保存结果resolutions = [1,2]for i, res in enumerate(resolutions): sc.tl.leiden(adata, resolution=res, key_added=f"leiden_res{res}")sc.pl.umap(adata, color="leiden_res2", legend_loc='on data',legend_fontsize=12) # 在数据点上显示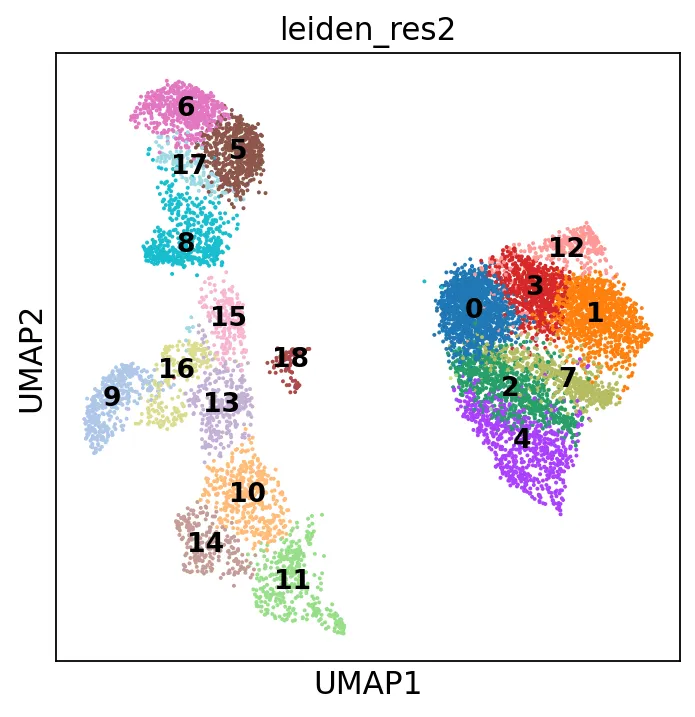
点图
通过点图对标记基因表达模式进行可视化验证
B_plasma_markers = { ct: [m for m in ct_markers if m in adata.var.index]for ct, ct_markers in marker_genes.items()if ct in B_plasma_cts}sc.pl.dotplot( adata, groupby="leiden_res2", var_names=B_plasma_markers, standard_scale="var", # standard scale: normalize each gene to range from 0 to 1)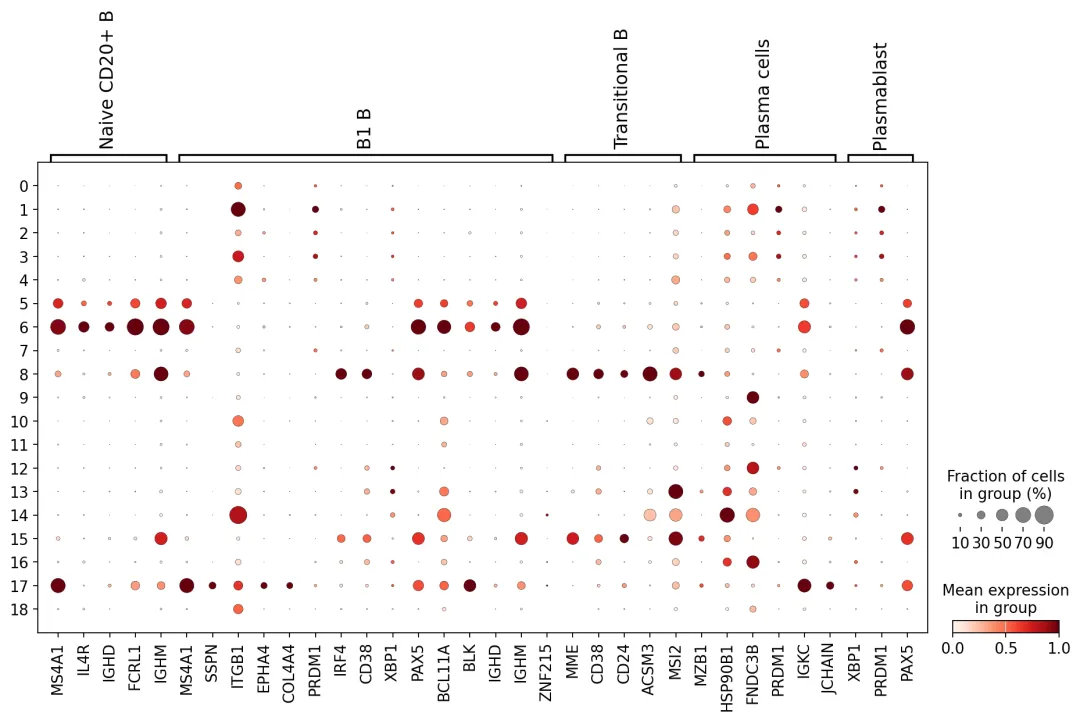
注释
结合UMAP图的直观观察与上述点图分析,我们现在可以开始对聚类进行注释:
B1 B细胞的注释较为困难,没有哪个聚类能表达所有B1 B标记基因,而多个聚类仅表达了部分标记。我们常发现适用于某个数据集的标记基因在其他数据集中效果不佳。这可能源于测序深度的差异,也可能因数据集或样本间的其他变异因素所致。
cl_annotation = {"5": "Naive CD20+ B","6": "Naive CD20+ B","8": "Transitional B","17": "B1 B", # note that IGHD and IGHM are negative markers, in this case more lowly expressed than in the other B cell clusters}adata.obs["manual_celltype_annotation"] = adata.obs.leiden_res2.map(cl_annotation)sc.pl.umap(adata, color=["manual_celltype_annotation"])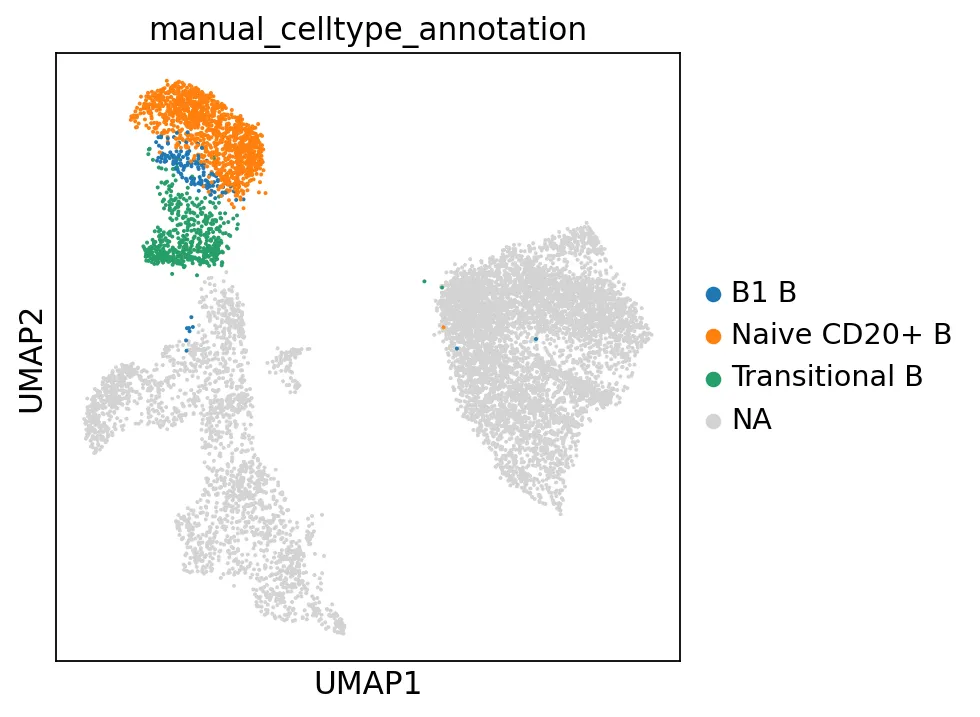
3.2 从聚类差异表达基因到聚类注释
相反地,我们可以计算每个聚类的差异表达基因,然后查证这些基因是否与已知的生物学特征(如细胞类型和/或状态)相关联。
计算每个聚类相对于adata中其余细胞的差异表达基因:
sc.tl.rank_genes_groups( adata, groupby="leiden_res2", method="wilcoxon", key_added="dea_leiden_2")# 过滤sc.tl.filter_rank_genes_groups( adata, min_in_group_fraction=0.2, max_out_group_fraction=0.2, key="dea_leiden_2", key_added="dea_leiden_2_filtered",)# 展示top5sc.pl.rank_genes_groups_dotplot( adata, groupby="leiden_res2", standard_scale="var", n_genes=5, key="dea_leiden_2_filtered",)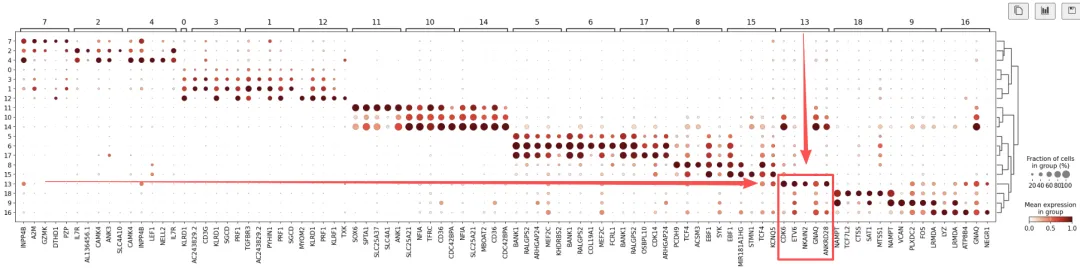
聚类13:
似乎拥有一组相对独特的标记基因,包括CDK6、ETV6、NKAIN2和GNAQ。通过检索发现,NKAIN2和ETV6是造血干细胞标记物[Shi等人, 2022][Wang等人, 1998](NKAIN2也出现在我们之前的列表中)。
从UMAP图中可以看到,这些基因在整个聚类13中均有表达:
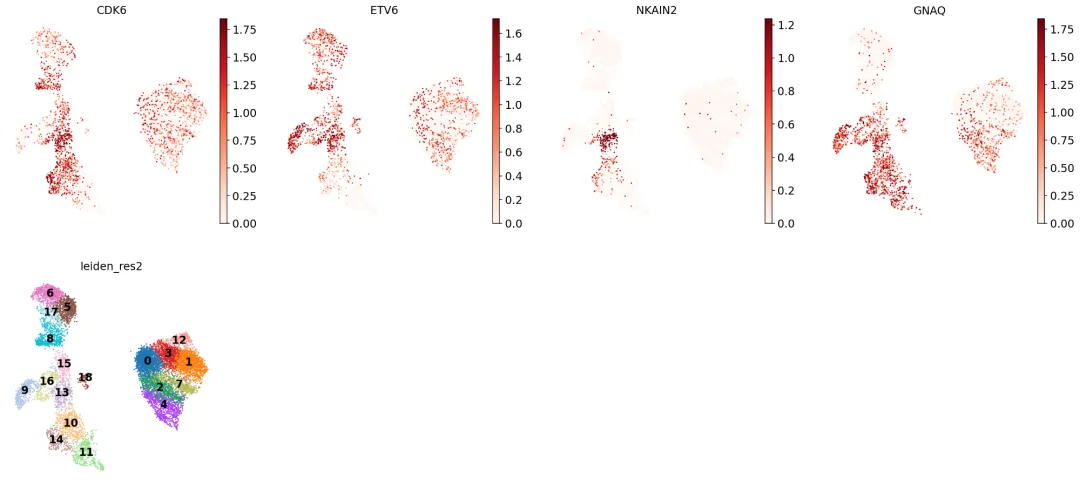
但是,观察巨核细胞/红细胞祖细胞("MK/E prog")的已知标记基因时,我们发现聚类13的部分细胞似乎属于该细胞类型:
sc.pl.umap( adata, color=[ "ZNF385D","ITGA2B","RYR3","PLCB1",], vmax="p99", legend_loc="on data", frameon=False, cmap="Reds",)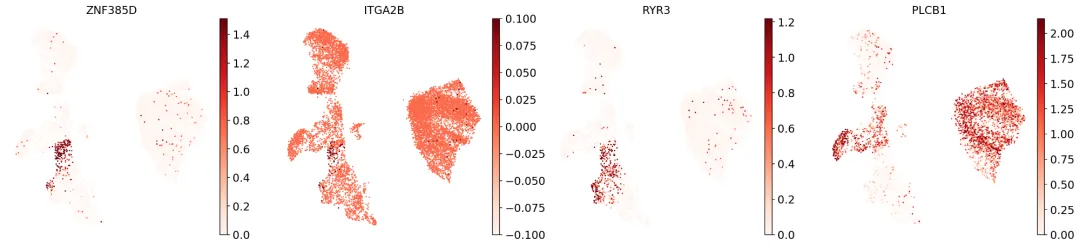
以上 两种方式凸显了基于标记基因注释的复杂性:它对所选聚类分辨率、所用标记基因集的稳健性和独特性,以及你对数据中预期细胞类型的了解程度均较为敏感。
最后保存一下数据:
cl_annotation["13"] = "HSCs + MK/E prog (?)"adata.obs["manual_celltype_annotation"] = adata.obs.leiden_res2.map(cl_annotation)下一篇我们来做基于自动化的数据注释。
转发:
生信入门&数据挖掘线上直播课2026年1月班,你的生物信息学入门课 时隔5年,我们的生信技能树VIP学徒继续招生啦 满足你生信分析计算需求的低价解决方案 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- Python学习【93】:学习Python用Pycharm、Jupyter还是Anaconda?
- 实战|Spring +Milvus,Java也能实现的企业级文档问答RAG
- 【播客】Kali Linux就业前景
- 【2026最新】100个Python常用语法,速记!
- 【已复现】多款 Linux telnetd服务中招,仅需一个参数即可远程获取 Root 权限,GNU Inetutils telnetd 远程认证绕过漏洞(CVE-2026-24061)
- 不写一行代码?保姆级教程:从0到1的Vibe Coding体验-前端篇
- Python 并发与 asyncio 3.6 优雅地关闭
- 码住收藏!25年新版计算机二级考试(Java语言程序设计)常考易错练习题及答案8
- 2025低代码5大核心趋势:AI原生、云边协同、垂直DSL崛起
- Maven4要来了:15年后,Java构建工具迎来“彻底重构”
