现在做RNA-seq分析,还要掌握Linux和R吗?
- 2026-06-24 11:03:50
本文由生物帮SA成员投稿。欢迎对某些内容感兴趣的朋友在文章下留言。
前期我们介绍了RNA-seq数据的分析流程[不会RNA-seq分析?一篇帮你理清所有步骤]。在具体分析时,很多朋友会卡在第一步:环境配置、命令行……然而在AI工具愈发强大的今天,做RNA-seq分析,AI能够帮助做什么呢?
🧠 原始数据处理
1. 这一阶段通常包括:质控(FastQC);比对(HISAT2/STAR);定量(featureCounts)。
2. 传统的处理方式包括:Linux命令行操作、shell脚本拼接跑流程。
3. AI可以帮助做什么:
①自动生成命令(例如比对和统计脚本)
②解释报错信息(如路径错误、依赖缺失)
③帮你整理pipeline思路(甚至生成Snakemake流程)
然而目前AI还不能完全替你“跑流程”。尤其是服务器环境、软件安装、路径配置等问题,仍然需要一定基础。
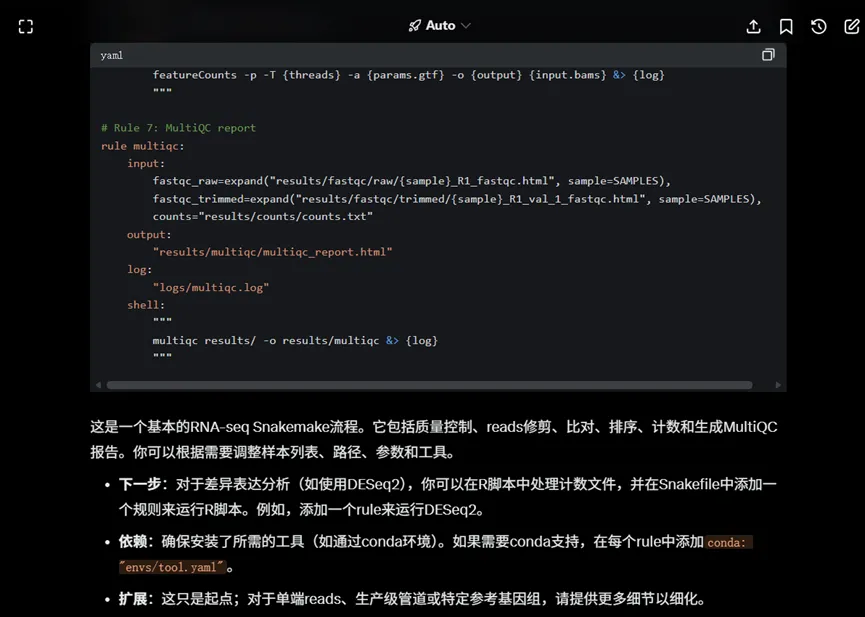
下图是Grok生成的RNA-seq流程,它包括质量控制、reads修剪、比对、排序、计数和生成MultiQC报告。
🌱 差异表达分析
1. 这一部分通常用R包完成,比如:DESeq2、edgeR。
2. AI的优势非常明显,例如:
①帮你写完整R代码(从读数据到画图)
②自动解释结果(log2FoldChange、padj等)
③提供参数建议(阈值如何设定)
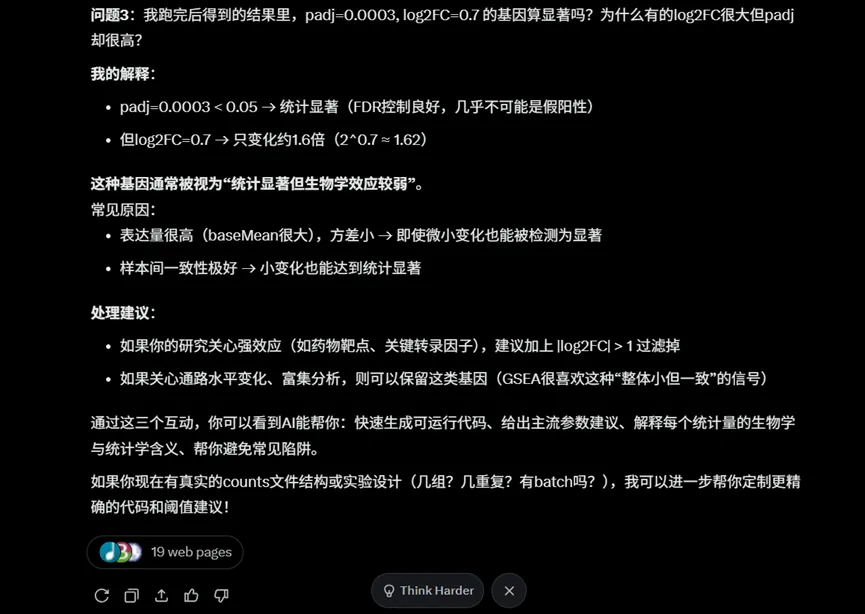
3. 如果你直接询问:padj < 0.05 和 |log2FC| > 1 合理吗?
AI不仅会回答,还会解释统计意义,这是Grok的回答结果:
🔬 功能富集分析
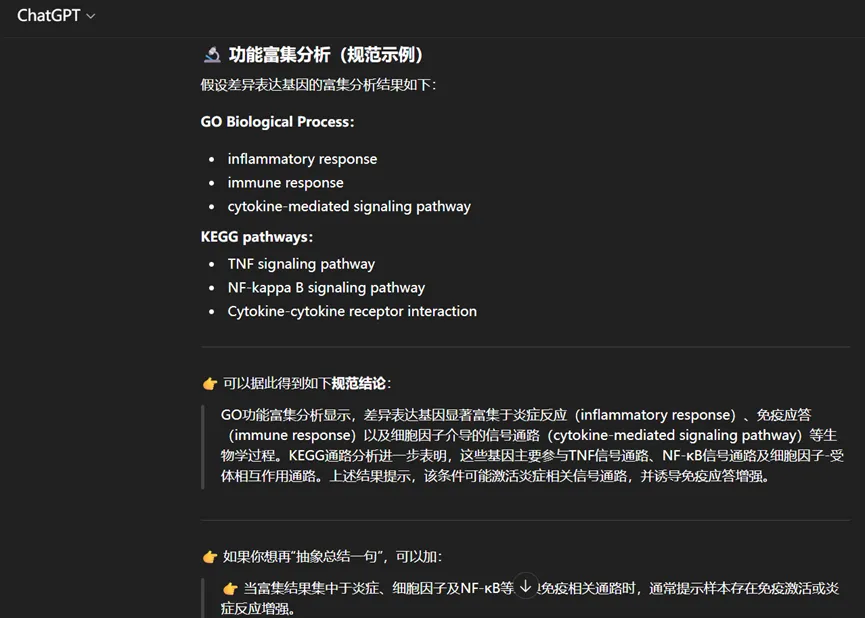
1. 对于传统的GO分析、KEGG通路分析,AI可以帮助总结通路意义、解释生物学背景等。
2. 输入一组富集通路,AI可以帮你总结:
“这些结果提示免疫反应被激活,尤其是炎症相关通路上调。”对写论文非常友好。
🦠 高级分析
1. 高级分析包括单细胞分析(Seurat/Scanpy)、基因调控网络(GRN)、虚拟敲除(如scTenifoldKnk)等。
以下是scTenifoldKnk的介绍界面:
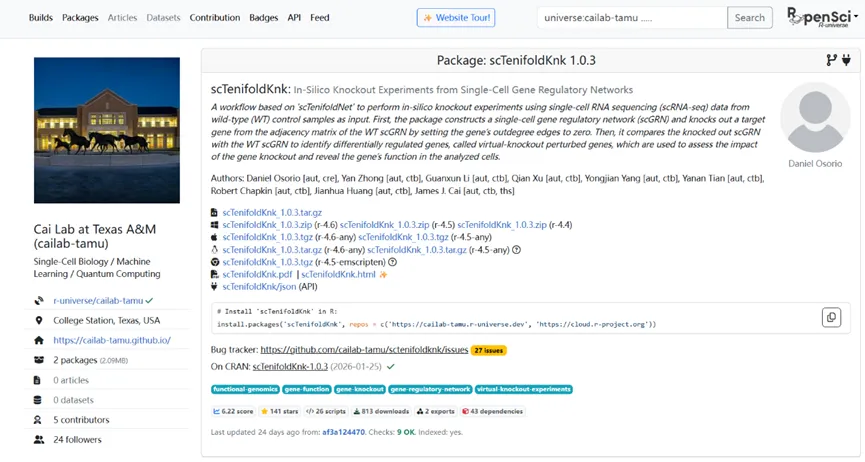
2. AI可以帮你理解方法、推荐工具、辅助解读结果,但目前仍然无法直接替你完成复杂分析、自动构建完整科研级pipeline。
主要原因是这些分析不仅是“技术问题”,更是“建模问题”。总体来说AI擅长“辅助理解和加速操作”,但还无法替代“完整分析能力”。
⚗️ 必须掌握编程语言吗
综合以上分析,对于普通的数据分析答案是非必须。
1. 选择现有分析工具
进行数据分析时可以选择一些在线分析工具,包括Galaxy(网页分析平台)、GEO自带分析工具(如GEO2R),以及一些商业或云端分析平台等。
AI在此可以发挥的作用主要包括:
①内容教学(例如“差异分析是指什么”);
②指导工具使用(参数怎么选、结果怎么看);
③解释输出结果(如log2FC、p值、生物意义)。
2. 编程分析
当你的数据量变大(几十几百G)、需要自定义分析流程、论文要求“可重复性”,建议掌握必要的编程技能。但此时AI可以帮助完成大部分的分析工作:
①自动生成代码(R / shell);
②帮助debug(解释报错原因);
③优化分析流程(推荐更合理方法);
④提供思路(该不该做批次校正)。

🌟关于生物帮SA
生物帮SA为公司旗下的生态交流平台,由清华大学博士团队(清华大学x-lab培育团队)创立,致力于打造学术一体化生态圈,全方位助力科研与职业发展:
[1] 解决生物问题:百余位来自清华、北大、Stanford University、University of Nebraska Lincoln、复旦、浙大、中科院等国内外顶尖高校的老师、工程师、博后、硕博生加入团队,提供科研问题解决方案,重点打造藻类合作、科研仪器定制、论文咨询、实验合作、知识产权等专项服务;
[2] 分享科研干货:涵盖生物知识、科研作图、经验交流,助力科研之路。
[3] 传播实用信息:分享就业资讯、试剂仪器信息和培训资源。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- Python Tkinter搭建工业流程动画?这活儿比你想的有意思多了!
- 中篇Python(ODOO)篇 第一章 Python基础(六)基础部分 2.基本数据类型
- 取代Maven!谷歌力荐这个Java构建工具!
- 用 Python 打造交易策略:3 个指标实现自动化交易回测
- 如何使用Python语言实现分布式自动化提升效率
- 春招_python基础_p19_continue_break_pass.py
- 因子投资实战(二):用Python挖掘Alpha因子
- 【计算机二级 Python・每日 10 考点】Day12
- Python:数值运算协议
- 用DeepSeeK怎么学习pythOn
