打开Ubuntu,输入 conda activate bioinfo,已进入环境,包会装到这个环境,且不污染系统
测试环境
退出python 可以输入exit()或者按ctrl+D,就会回到linux
练习:运用python画一张基因表达图,读取一个RNA-seq表达矩阵并画图
第一步退出python交互模式
第二步,安装今天要用的包
在Ubuntu输入conda install pandas numpy matplotlib seaborn jupyter -y
conda :包管理器+环境管理器
conda install :用conda安装软件
pandas :数据处理库,读取数据表及处理矩阵,统计,清洗数据
numpy:科学计算库,用于数值计算,矩阵运算,数组
matplotlib :绘图库,画折线图,散点图,箱线图,热图
seaborn :高级绘图库,基于matplotlib,图更加漂亮,统计图更方便
jupyter :交互式编程环境,写代码,运行代码,显示图,写笔记
-y yes
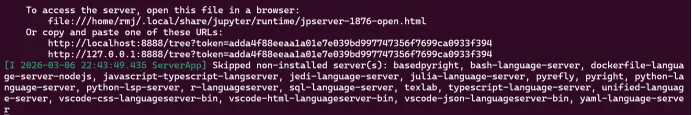
第三步,输入jupyter notebook --no-browser
jupyter notebook 启动jupyter notebook服务器,即启动一个本地网页服务,可以在浏览器里写python,即jupyter实际上是python+浏览器的工作环境
no-browser:不要自动打开浏览器,因为目前在windows+WSL Ubuntu,如果不用这个参数,jupyter会尝试用linux浏览器打开,但是wsl没有浏览器,可能会报错
运行完后,复制网址地址至Edge浏览器打开,进入notebook,代码在linux运行,界面在windows浏览器显示
在new下面的python3打开,即看到写代码的地方
小型生信任务:读取数据,查看数据,画科研图
在jupyter notebook输入
首先输入
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
解读:import pandas as pd :把pandas工具库加载到python里使用,且起个简称pd,以后用的时候直接写pd.read_csv()
其余同理
接着输入:
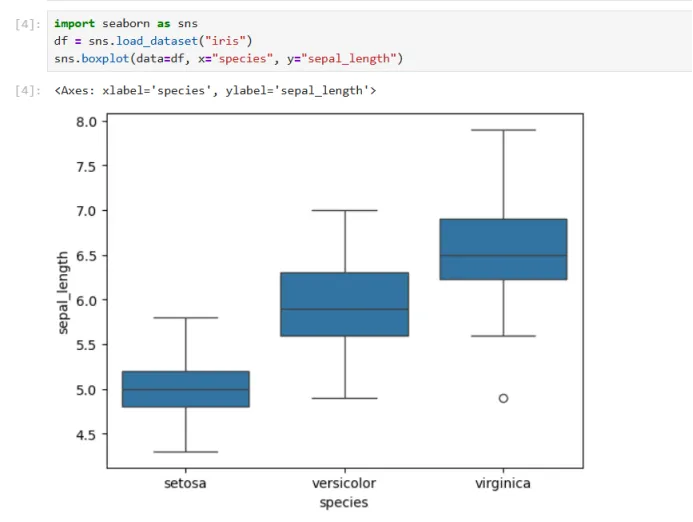
import seaborn as sns
df = sns.load_dataset("iris")
sns.boxplot(data=df, x="species", y="sepal_length")
代码解读:
import seaborn as sns:导入seaborn这个绘图库,并简称为sns,以后调用这个数据库就是sns.heatmap( ) , sns.scatterplot( ) , sns.boxplot( )
df = sns.load_dataset("iris") : sns.load_dataset(“iris”)是seaborn自带的一个示例数据集,iris是一个经典的生物学数据,这一部分的意思是把数据存到变量df里,df通常代表DataFrame(数据表),总得意思就是把iris数据集读进来,存成一个数据表df
sns.boxplot(data=df, x="species", y="sepal_length")
用seaborn画一个箱线图(boxplot),x轴是花的种类,y轴是花萼长度
第一步,确保导入库,先运行
import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
第二步,创建一个模拟的RNA-seq数据
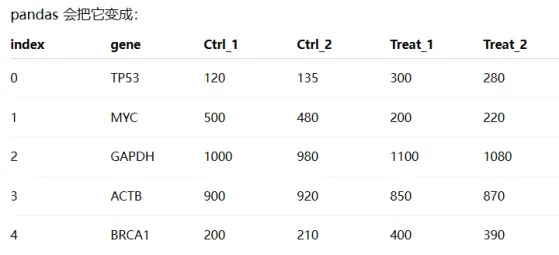
data = {
"gene": ["TP53", "MYC", "GAPDH", "ACTB", "BRCA1"],
"Ctrl_1": [120, 500, 1000, 900, 200],
"Ctrl_2": [135, 480, 980, 920, 210],
"Treat_1": [300, 200, 1100, 850, 400],
"Treat_2": [280, 220, 1080, 870, 390]
}
df = pd.DataFrame(data)
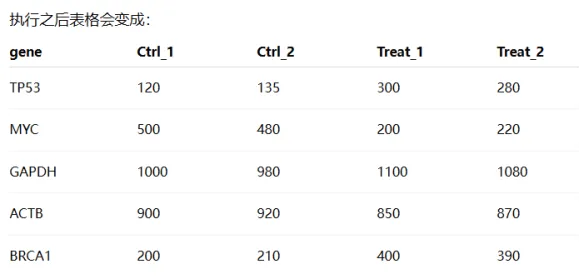
df = df.set_index("gene")
代码解读:df =pd.DataFrame(data),把data这个python 字典转换成表格数据(DataFrame)
DataFrame:类似Excel的表格结构
df =df.set_index(“gene”):是把gene这一列变成行标签index
原因:表达矩阵的标准格式就是gene×sample
第三步,做log转换:让数据更容易画图
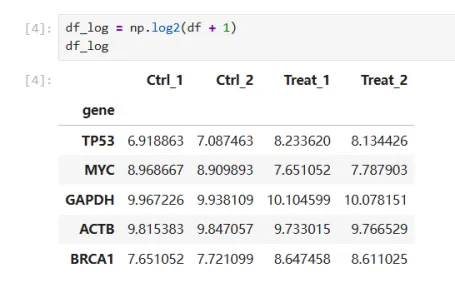
df_log = np.log2(df + 1)
df_log
代码解读:df_log = np.log2(df + 1),给所有表达量加1,再做log2转换
选择log2的原因是RNA-seq表达量分布非常不均匀,做了log2转换后,差距不会那么夸张,加1是因为如果表达量为0,log2(0)是不存在的。这也是为什么论文中热图颜色范围大多是-2 -1 0 1 2 ,而不是原始表达量
df_log:显示转换后的表达矩阵
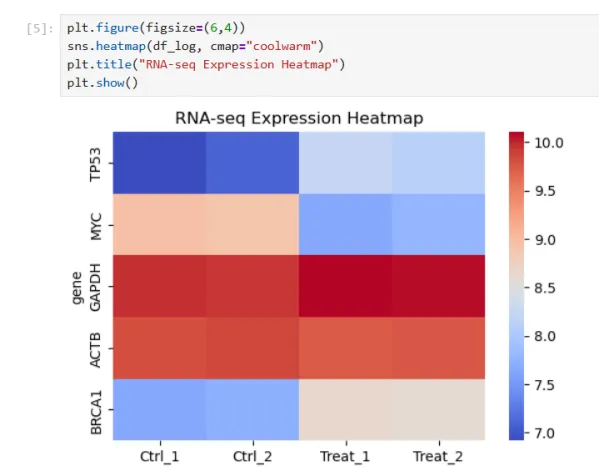
第四步,画热图
plt.figure(figsize=(6,4))
sns.heatmap(df_log, cmap="coolwarm")
plt.title("RNA-seq Expression Heatmap")
plt.show()
代码解读:plt.figure(figsize=(6,4)) 创建一个画布(图像窗口),plt是matplotlib画图工具的简称,figure()创建一张新的图,figsize=(6,4)图像大小是宽=6,高=4,单位是英寸(inch)
sns.heatmap(df_log, cmap="coolwarm"):sns是统计绘图库,heatmap是热图,不同数值代表不同颜色,画出颜色矩阵,df-log=np.log2(df+1),热图用的是log2表达量,cmap="coolwarm"是颜色方案,coolwarm表示蓝色-低表达,白色-中等,红色-高表达,其余常见配色有viridis, magma, plasma, RdBu
plt.title("RNA-seq Expression Heatmap"):绘图添加标题
plt.show():把图真正显示出来
真正的RNA-seq热图会多两步,即Z-score标准化,不同基因表达范围统一,第二就是聚类(基因聚类,样本聚类)
结果展示:
插曲:
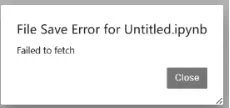
遇到的一个 Jupyter Notebook 的常见 WSL 问题:
File Save Error for Untitled.ipynb Failed to fetch,意思是浏览器和 Jupyter 服务器的连接 断开了,所以 Notebook 无法保存,也无法正常运行代码。这不是代码问题,而是 Jupyter 会话断开了。
解决办法:回到Ubuntu终端,先按ctrl+C,Shutdown this notebook server (y/[n])?,输入y,然后回车,再重启jupyter,在终端输入jupyter notebook ,再重新打开网页,新建一个notebook,继续运行代码,更稳定的习惯jupyter notebook --no-browser --port=8888,重新关掉打开也行。






 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
