R+Python组合拳可能是分析SCENIC最好的方式
- 2026-07-04 02:37:00
一、写在前面
在利用SCENIC进行转录因子共表达网络预测时有一步非常头疼的runGenie3,这一步基于随机森林算法推断基因调控网络(GRN),输出结果包含TF-基因对及其调控强度的矩阵。这一步不仅耗费很多内存,并且还不支持在R中进行并行计算,通常1W个细胞的计算时间就能达到一周以上,甚至经常运行到一半R崩溃了,显然不能满足大家日常生产的需求。但在python中的grnboost2可以利用分布式并行计算K可以大大缩短这一过程的计算时间:比R版本快几十倍| Pyscenic单细胞转录因子预测,毛病就是PySCENIC会略去很多SCENIC的中间输出结果与可视化,这显然是不能被接受的 。在SCENIC单细胞转录因子预测学习手册的教程中我们也提到过加速方法:,可以直接在R中利用reticulate包调用python库arboreto.algo来完成grnboost2的计算过程。当然,如果实在觉得学习困难,也别难为自己,欢迎找我们代劳~联系客服微信[Biomamba_zhushou]留下您的需求。
# 基因共表达网络计算mymethod <-'grnboost2'# 'grnboost2'library(reticulate)if(mymethod=='runGenie3'){runGenie3(exprMat_filtered_log, scenicOptions)}else{ arb.algo =import('arboreto.algo') tf_names =getDbTfs(scenicOptions) tf_names = Seurat::CaseMatch(search = tf_names,match =rownames(exprMat_filtered)) adj = arb.algo$grnboost2(as.data.frame(t(as.matrix(exprMat_filtered))),tf_names=tf_names, seed=2025L )colnames(adj) =c('TF','Target','weight')saveRDS(adj,file=getIntName(scenicOptions,'genie3ll'))}问题是,上面的这个R代码框经常会遇到arboreto加载失败,要么找不到: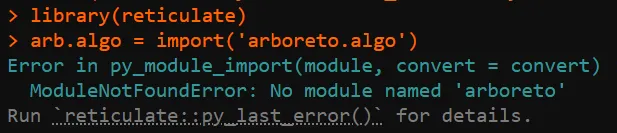
要么依赖库出问题
reticulate::use_python('/home/biomamba/.local/share/r-miniconda/envs/r-reticulate/bin/python') > arb.algo = import('arboreto.algo') Error in py_module_import(module, convert = convert) : ImportError: /usr/lib/x86_64-linux-gnu/libstdc++.so.6: version `GLIBCXX_3.4.29' not found (required by /home/biomamba/.local/share/r-miniconda/envs/r-reticulate/lib/python3.8/site-packages/pandas/_libs/window/aggregations.cpython-38-x86_64-linux-gnu.so) Run `reticulate::py_last_error()` for details.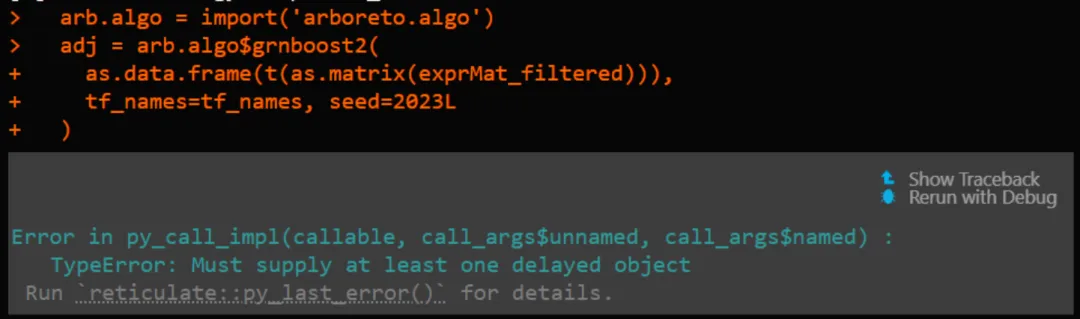
所以,最稳妥的办法是R和Python各忙各的,即生成exprMat_filtered后导出到本地,在纯粹的Python中完成。可视化集锦如下:
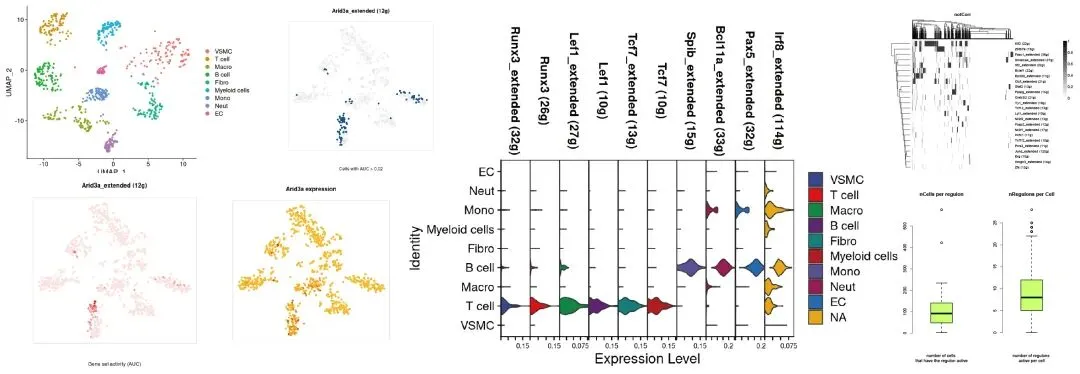
二、R+Python组合拳
如果以下内容有理解困难的地方,可先行学习:SCENIC单细胞转录因子 学习手册
1加载R包
if (!requireNamespace("BiocManager", quietly =TRUE))install.packages("BiocManager")BiocManager::version()## [1] '3.20'#当你的bioconductor版本大于4.0时if(!require(SCENIC))BiocManager::install(c("AUCell", "RcisTarget"),ask = F,update = F);BiocManager::install(c("GENIE3"),ask = F,update = F)#这三个包显然是必须安装的### 可选的包:#AUCell依赖包if(!require(SCENIC))BiocManager::install(c("zoo", "mixtools", "rbokeh"),ask = F,update = F) ###t-SNEs计算依赖包:if(!require(SCENIC))BiocManager::install(c("DT", "NMF", "ComplexHeatmap", "R2HTML", "Rtsne"),ask = F,update = F)# #这几个包用于并行计算,很遗憾,Windows下并不支持,所以做大量数据计算时最好转战linux:if(!require(SCENIC))BiocManager::install(c("doMC", "doRNG"),ask = F,update = F)#可视化输出# To export/visualize in http://scope.aertslab.orgif (!requireNamespace("devtools", quietly =TRUE))install.packages("devtools")if(!require(SCopeLoomR))devtools::install_github("aertslab/SCopeLoomR", build_vignettes =TRUE)#SCopeLoomR用于获取测试数据if (!requireNamespace("arrow", quietly =TRUE)) BiocManager::install('arrow')#这个包不装上在runSCENIC_2_createRegulons那一步会报错,提示'dbs'不存在library(SCENIC)#这就安装好了,试试能不能正常加载if(!require(SCENIC))devtools::install_github("aertslab/SCENIC") packageVersion("SCENIC")#这里我安装的是1.3.1## [1] '1.3.1'library(SCENIC)library(RcisTarget)library(AUCell)library(Seurat)2、R中的预处理
# 初始化SCNIE 设置data(list="motifAnnotations_mgi_v9", package="RcisTarget")motifAnnotations_mgi <- motifAnnotations_mgi_v9scenicOptions <-initializeScenic(org="mgi",#mouse填'mgi', human填'hgnc',fly填'dmel') dbDir="./data/mouse.mm9/", nCores=6)#这里可以设置并行计算## Motif databases selected: ## mm9-500bp-upstream-7species.mc9nr.feather ## mm9-tss-centered-10kb-7species.mc9nr.feather## Using the column 'features' as feature index for the ranking database.## Using the column 'features' as feature index for the ranking database.library(Seurat)# 读取测试对象scRNA <-readRDS('./data/pbmcrenamed.rds') # 查看测试对象降维图:DimPlot(scRNA) 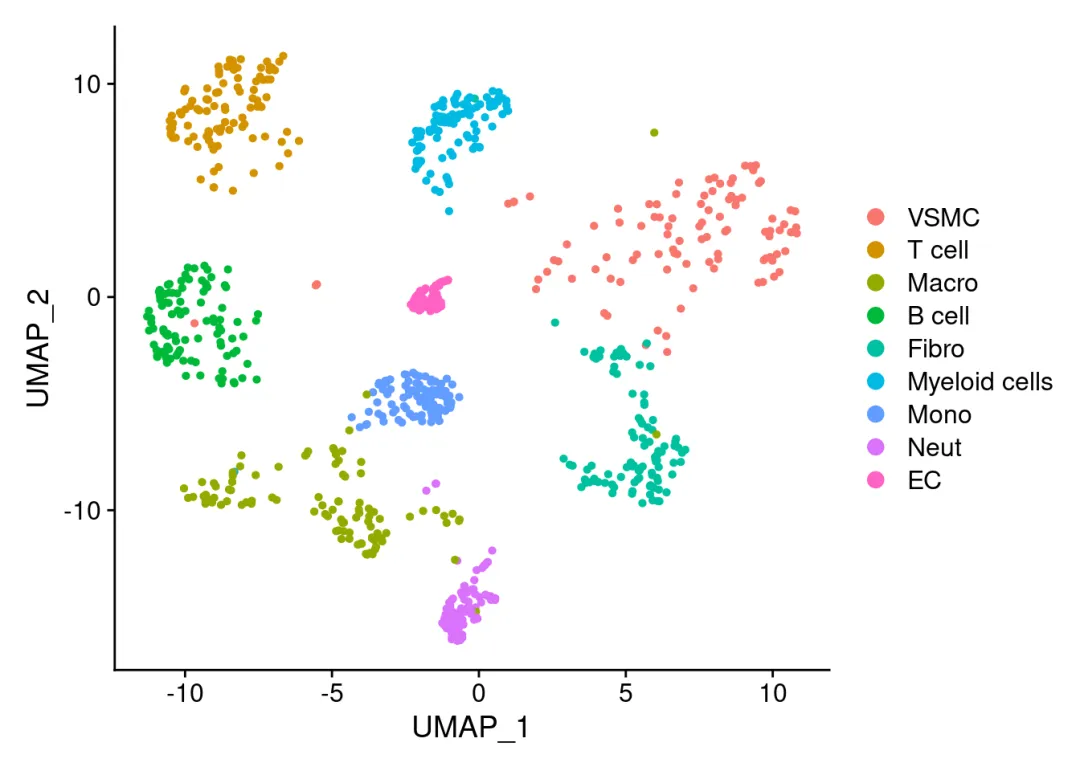
# 获取表达矩阵:exprMat <-GetAssayData(scRNA ,'RNA','count')# 转换为矩阵对象:exprMat <-as.matrix(exprMat)# 初步过率基因:genesKept <-geneFiltering(exprMat, scenicOptions)## Maximum value in the expression matrix: 26404## Ratio of detected vs non-detected: 0.12## Number of counts (in the dataset units) per gene:## Min. 1st Qu. Median Mean 3rd Qu. Max. ## 0.0 3.0 48.0 407.6 214.0 187278.0## Number of cells in which each gene is detected:## Min. 1st Qu. Median Mean 3rd Qu. Max. ## 0.00 3.00 39.00 99.42 143.00 897.00## ## Number of genes left after applying the following filters (sequential):## 11369 genes with counts per gene > 27## 11338 genes detected in more than 9 cells## Using the column 'features' as feature index for the ranking database.## 10021 genes available in RcisTarget database## Gene list saved in int/1.1_genesKept.RdsexprMat_filtered <- exprMat[genesKept, ]# 计算spearman相关性: runCorrelation(exprMat_filtered, scenicOptions)# 取log:exprMat_filtered_log <-log2(exprMat_filtered+1) # 获取TF的名称:tf_names =getDbTfs(scenicOptions)tf_names = Seurat::CaseMatch(search = tf_names,match =rownames(exprMat_filtered))# 导出表达矩阵(需要转置,因为Python中通常是行为样本,列为基因)便于后续Python读取:write.csv(t(exprMat_filtered), file ="./data/expr_matrix_transposed.csv", row.names =TRUE)# 导出TF名称列表write.csv(data.frame(tf_names = tf_names), file ="./data/tf_names.csv", row.names =FALSE)3、Python中完成grnboost
### 以下命令在Python中执行 ###import pandas as pdfrom arboreto.algo import grnboost2# 读取R导出的数据expr_matrix = pd.read_csv("data/expr_matrix_transposed.csv", index_col=0)tf_names = pd.read_csv("data/tf_names.csv")["tf_names"].tolist()# 确保TF名称在表达矩阵的基因中存在available_genes = expr_matrix.columns.tolist()tf_names = [tf for tf in tf_names if tf in available_genes]# 运行GRNBoost2adj = grnboost2( expr_matrix, tf_names=tf_names, seed=2023, verbose=True)# 重命名列以匹配R中的预期格式adj.columns = ['TF', 'Target', 'weight']# 保存结果(R可以读取的CSV格式)adj.to_csv("data/grnboost2_results.csv", index=False)以下是运行提示信息:
>>>### 以下命令在Python中执行 ###>>>import pandas as pd>>>from arboreto.algo import grnboost2>>>>>># 读取R导出的数据>>> expr_matrix = pd.read_csv("data/expr_matrix_transposed.csv", index_col=0)>>> tf_names = pd.read_csv("data/tf_names.csv")["tf_names"].tolist()>>>>>># 确保TF名称在表达矩阵的基因中存在>>> available_genes = expr_matrix.columns.tolist()>>> tf_names = [tf for tf in tf_names if tf in available_genes]>>>>>># 运行GRNBoost2>>> adj = grnboost2(... expr_matrix,... tf_names=tf_names,... seed=2023,... verbose=True... )preparing dask clientparsing inputcreating dask graph19 partitionscomputing dask graphshutting down client and local clusterfinished>>>>>># 重命名列以匹配R中的预期格式>>> adj.columns = ['TF', 'Target', 'weight']>>>>>># 保存结果(R可以读取的CSV格式)>>> adj.to_csv("data/grnboost2_results.csv", index=False)4、R语言完成SCENIC的经典4步骤
回到R语言中:
# 读取Python输出的GRN结果adj <-read.csv("data/grnboost2_results.csv")# 转换为数据框并设置列名(确保与之前的格式一致)adj <-as.data.frame(adj)colnames(adj) <-c('TF', 'Target', 'weight')# 保存为RDS文件,供后续分析使用saveRDS(adj, file =getIntName(scenicOptions, 'genie3ll'))# 验证结果cat("成功读取GRNBoost2结果,共", nrow(adj), "条调控关系\n")# 运行经典的SCENIC step1-4exprMat_log <-log2(exprMat+1)scenicOptions@settings$dbs <- scenicOptions@settings$dbs["10kb"] # Toy run settingsscenicOptions <-runSCENIC_1_coexNetwork2modules(scenicOptions)scenicOptions <-runSCENIC_2_createRegulons(scenicOptions, coexMethod=c("top5perTarget")) # Toy run settingsscenicOptions <-runSCENIC_3_scoreCells(scenicOptions, exprMat_log)scenicOptions <-runSCENIC_4_aucell_binarize(scenicOptions)#将AUCell矩阵二元化# 保存镜像save.image('data/biomamba_scenic.rdata')# 这时我们就拥有与纯在R语言中执行SCENIC一样的工程文件:tree ./int## [01;34m./int[00m## ├── 1.1_genesKept.Rds## ├── 1.2_corrMat.Rds## ├── 1.4_GENIE3_linkList.Rds## ├── 1.5_weightPlot.pdf## ├── 1.6_tfModules_asDF.Rds## ├── 2.1_tfModules_forMotifEnrichmet.Rds## ├── 2.2_motifs_AUC.Rds## ├── 2.3_motifEnrichment.Rds## ├── 2.4_motifEnrichment_selfMotifs_wGenes.Rds## ├── 2.5_regulonTargetsInfo.Rds## ├── 2.6_regulons_asGeneSet.Rds## ├── 2.6_regulons_asIncidMat.Rds## ├── 3.1_regulons_forAUCell.Rds## ├── 3.2_aucellGenesStats.pdf## ├── 3.3_aucellRankings.Rds## ├── 3.4_regulonAUC.Rds## ├── 3.5_AUCellThresholds_Info.tsv## ├── 3.5_AUCellThresholds.Rds## ├── 4.1_binaryRegulonActivity.Rds## ├── 4.2_binaryRegulonActivity_nonDupl.Rds## ├── 4.3_regulonSelections.Rds## ├── 4.4_binaryRegulonOrder.Rds## └── tSNE_AUC_50pcs_30perpl.Rds## ## 0 directories, 23 files# 读取AUCell Score矩阵:auc_score <-getAUC(readRDS("int/3.4_regulonAUC.Rds"))# 添加到注释信息中:scRNA <-AddMetaData(scRNA,metadata =t(auc_score))# 小提琴挑几个转录因子瞅瞅:VlnPlot(scRNA,rownames(auc_score)[1:10],stack = T,cols =c(ggsci::pal_aaas()(8),ggsci::pal_bmj()(8)) )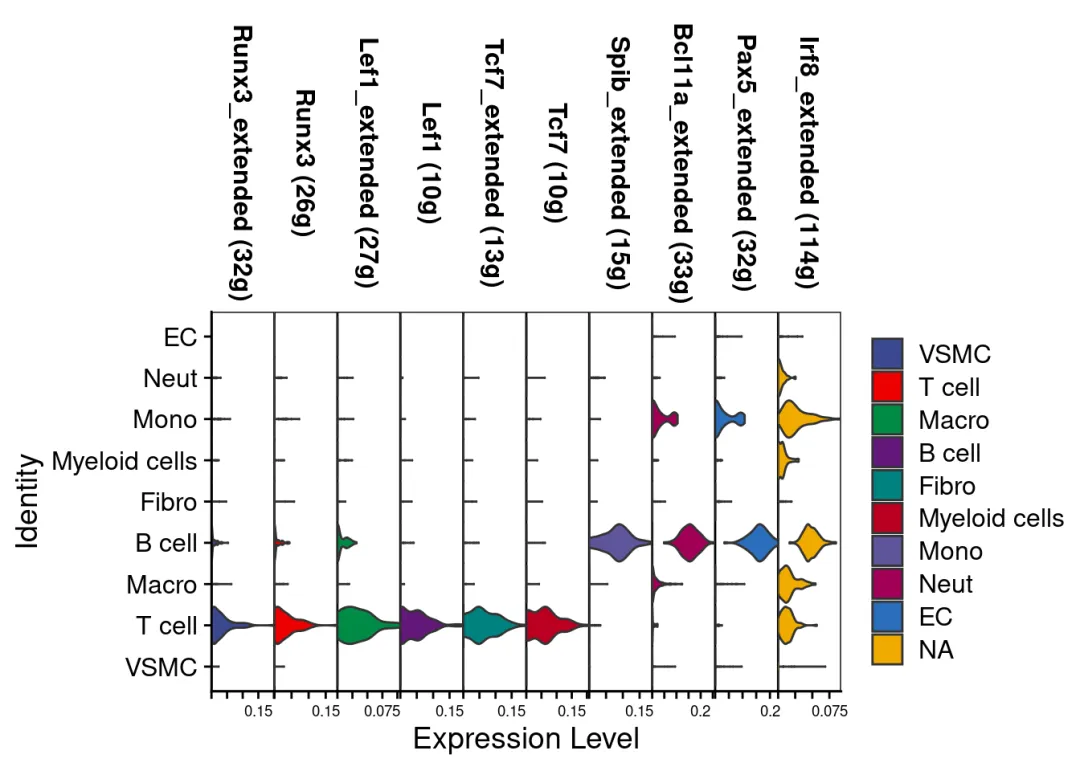
三、测试文件与演示环境
通过网盘分享的文件:R与Python组合分析SCENIC流程
链接: https://pan.baidu.com/s/1DQdzV7QzuwJ89-Cs7xsnmQ?pwd=tfdx
提取码: tfdx


sessionInfo()## R version 4.4.2 (2024-10-31)## Platform: x86_64-pc-linux-gnu## Running under: Ubuntu 20.04.4 LTS## ## Matrix products: default## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3 ## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0## ## locale:## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C ## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8 ## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 ## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C ## [9] LC_ADDRESS=C LC_TELEPHONE=C ## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C ## ## time zone: Etc/UTC## tzcode source: system (glibc)## ## attached base packages:## [1] stats graphics grDevices utils datasets methods base ## ## other attached packages:## [1] Seurat_5.2.1 SeuratObject_5.0.2 sp_2.2-0 AUCell_1.28.0 ## [5] RcisTarget_1.26.0 SCopeLoomR_0.13.0 SCENIC_1.3.1 ## ## loaded via a namespace (and not attached):## [1] RcppAnnoy_0.0.22 splines_4.4.2 ## [3] later_1.4.1 bitops_1.0-9 ## [5] tibble_3.2.1 R.oo_1.27.0 ## [7] polyclip_1.10-7 graph_1.84.1 ## [9] XML_3.99-0.18 fastDummies_1.7.5 ## [11] lifecycle_1.0.4 globals_0.16.3 ## [13] lattice_0.22-6 hdf5r_1.3.12 ## [15] MASS_7.3-64 magrittr_2.0.3 ## [17] plotly_4.10.4 sass_0.4.9 ## [19] rmarkdown_2.29 jquerylib_0.1.4 ## [21] yaml_2.3.10 remotes_2.5.0 ## [23] httpuv_1.6.15 sctransform_0.4.1 ## [25] spam_2.11-1 spatstat.sparse_3.1-0 ## [27] sessioninfo_1.2.2 pkgbuild_1.4.6 ## [29] reticulate_1.43.0.9001 cowplot_1.1.3 ## [31] pbapply_1.7-2 DBI_1.2.3 ## [33] RColorBrewer_1.1-3 abind_1.4-8 ## [35] pkgload_1.4.0 zlibbioc_1.52.0 ## [37] Rtsne_0.17 GenomicRanges_1.58.0 ## [39] purrr_1.0.2 R.utils_2.12.3 ## [41] BiocGenerics_0.52.0 RCurl_1.98-1.16 ## [43] GenomeInfoDbData_1.2.13 IRanges_2.40.1 ## [45] S4Vectors_0.44.0 ggrepel_0.9.6 ## [47] irlba_2.3.5.1 spatstat.utils_3.1-4 ## [49] listenv_0.9.1 openintro_2.5.0 ## [51] goftest_1.2-3 airports_0.1.0 ## [53] RSpectra_0.16-2 spatstat.random_3.4-1 ## [55] annotate_1.84.0 fitdistrplus_1.2-2 ## [57] parallelly_1.42.0 DelayedMatrixStats_1.28.1 ## [59] codetools_0.2-20 DelayedArray_0.32.0 ## [61] tidyselect_1.2.1 UCSC.utils_1.2.0 ## [63] farver_2.1.2 spatstat.explore_3.4-3 ## [65] matrixStats_1.5.0 stats4_4.4.2 ## [67] jsonlite_1.8.9 ellipsis_0.3.2 ## [69] progressr_0.15.1 ggridges_0.5.6 ## [71] survival_3.8-3 tools_4.4.2 ## [73] ica_1.0-3 Rcpp_1.0.14 ## [75] glue_1.8.0 gridExtra_2.3 ## [77] SparseArray_1.6.0 xfun_0.50 ## [79] MatrixGenerics_1.18.1 usethis_3.1.0 ## [81] GenomeInfoDb_1.42.1 dplyr_1.1.4 ## [83] withr_3.0.2 BiocManager_1.30.25 ## [85] fastmap_1.2.0 digest_0.6.37 ## [87] R6_2.5.1 mime_0.12 ## [89] colorspace_2.1-1 scattermore_1.2 ## [91] tensor_1.5 spatstat.data_3.1-6 ## [93] RSQLite_2.3.9 R.methodsS3_1.8.2 ## [95] ggsci_3.2.0 tidyr_1.3.1 ## [97] generics_0.1.3 data.table_1.16.4 ## [99] usdata_0.3.1 httr_1.4.7 ## [101] htmlwidgets_1.6.4 S4Arrays_1.6.0 ## [103] uwot_0.2.2 pkgconfig_2.0.3 ## [105] gtable_0.3.6 blob_1.2.4 ## [107] lmtest_0.9-40 XVector_0.46.0 ## [109] htmltools_0.5.8.1 profvis_0.4.0 ## [111] dotCall64_1.2 GSEABase_1.64.0 ## [113] scales_1.3.0 Biobase_2.66.0 ## [115] png_0.1-8 spatstat.univar_3.1-3 ## [117] knitr_1.49 rstudioapi_0.17.1 ## [119] reshape2_1.4.4 tzdb_0.4.0 ## [121] nlme_3.1-168 cachem_1.1.0 ## [123] zoo_1.8-12 stringr_1.5.1 ## [125] KernSmooth_2.23-26 parallel_4.4.2 ## [127] miniUI_0.1.1.1 arrow_18.1.0.1 ## [129] AnnotationDbi_1.68.0 pillar_1.10.1 ## [131] grid_4.4.2 vctrs_0.6.5 ## [133] RANN_2.6.2 urlchecker_1.0.1 ## [135] promises_1.3.2 xtable_1.8-4 ## [137] cluster_2.1.8 evaluate_1.0.3 ## [139] readr_2.1.5 cli_3.6.3 ## [141] compiler_4.4.2 rlang_1.1.5 ## [143] crayon_1.5.3 future.apply_1.11.3 ## [145] labeling_0.4.3 plyr_1.8.9 ## [147] fs_1.6.5 stringi_1.8.4 ## [149] deldir_2.0-4 viridisLite_0.4.2 ## [151] assertthat_0.2.1 munsell_0.5.1 ## [153] Biostrings_2.74.1 lazyeval_0.2.2 ## [155] spatstat.geom_3.4-1 devtools_2.4.5 ## [157] Matrix_1.7-2 RcppHNSW_0.6.0 ## [159] hms_1.1.3 patchwork_1.3.0 ## [161] sparseMatrixStats_1.18.0 bit64_4.6.0-1 ## [163] future_1.34.0 ggplot2_3.5.1 ## [165] KEGGREST_1.46.0 shiny_1.10.0 ## [167] SummarizedExperiment_1.36.0 ROCR_1.0-11 ## [169] igraph_2.1.4 memoise_2.0.1 ## [171] bslib_0.9.0 bit_4.5.0.1 ## [173] cherryblossom_0.1.0

如何联系我们


已有生信基地联系方式的同学无需重复添加



