python 单细胞流程-质控
- 2026-07-02 16:41:31
为什么需要数据清洗与质控?
在单细胞测序数据中,基于最常见的 10X Genomics 的“油包水”微体系,会出现多个质量问题。
低质量/死亡细胞:死亡或破裂细胞常表现为基因数低、UMI 数低、线粒体基因比例高。这类细胞的表达谱更多反映 RNA 降解或应激,而不是正常生物状态。 空液滴和环境 RNA:droplet-based scRNA-seq 里有些 barcode 不是细胞,而是空液滴或环境 RNA 污染。如果保留,会产生虚假的细胞群或假阳性 marker。 双细胞/多细胞 doublets:一个液滴里捕获了两个或多个细胞,会形成“混合表达谱”。这可能被误判成新的细胞亚群,尤其在聚类时很危险。 测序深度和文库复杂度差异:不同细胞捕获到的 RNA 数量差别很大。低复杂度细胞会导致 dropout 多、表达不稳定;高 UMI/高基因数也可能提示 doublet。 批次效应和样本处理差异:解离时间、组织来源、建库批次、测序深度都会影响表达矩阵。
但这些质量问题并不是必须强制每个都需要过滤,我们只需要解决主要干扰即可。低质量/死亡细胞无疑是过滤的首要关注。
读取数据
本分析使用 scanpy 内置的 pbmc3k 示例数据集。该数据集来源于 10x Genomics 平台测序的人外周血单个核细胞(Peripheral Blood Mononuclear Cells, PBMCs),包含约 3,000 个细胞的单细胞转录组表达数据。
通过 adata = sc.datasets.pbmc3k() 可以直接加载该数据集,并以 AnnData 对象的形式进行后续分析。
import scanpy as sc
adata = sc.datasets.pbmc3k()
print(adata.shape) # 细胞数 x 基因数
print(adata.obs.head()) # 细胞信息
print(adata.var.head()) # 基因信息
低质量/死亡细胞过滤
在单细胞转录组数据分析中,低质量细胞或死亡细胞会引入明显噪音,影响后续聚类、差异表达分析和细胞类型注释。这类细胞常因细胞膜受损导致胞质 mRNA 泄漏,因此通常表现为测序深度较低、检测到的基因数量较少,同时线粒体基因 reads 占比较高。质控时可根据这些 QC 指标设定过滤阈值,去除明显异常的细胞。常用做法是联合考察每个细胞的总 UMI/read 数、检测基因数和线粒体基因比例,例如过滤掉基因数过低、总计数过低或线粒体比例过高的细胞。对于较大数据集,也可采用基于 MAD 的自动阈值方法,将偏离中位数超过一定倍数 MAD 的细胞标记为异常值。需要注意的是,过滤标准不宜过严,应结合多个指标综合判断,以避免误删真实存在的活细胞群或稀有细胞亚群。
计算细胞指标
在进行细胞质控时,第一步是计算各类 QC 指标。通常可使用 scanpy中的 sc.pp.calculate_qc_metrics 函数,该函数不仅可以计算每个细胞的总计数、检测基因数等基础质控指标,还可以统计特定基因类别在总表达量中的占比。因此,在计算前需要先定义线粒体基因、核糖体基因和血红蛋白基因等基因集合。
需要注意的是,不同物种的线粒体基因命名规则不同:在人类数据中,线粒体基因通常以 MT- 为前缀;而在小鼠数据中,通常以小写的 mt-为前缀。
不同数据集也会有不同的数据特征,可进一步根据数据特征综合判断。核糖体蛋白基因比例高,可能说明细胞处于较强的蛋白质合成状态,也可能反映某些细胞类型本身具有较高翻译活性。因此它不一定代表低质量细胞,但可以作为判断细胞状态和批次差异的辅助指标。如果某一批样本中核糖体比例整体异常偏高,可能提示样本处理或测序质量存在差异。 血红蛋白基因比例高,常见于血液或骨髓样本中,可能提示红细胞或红系细胞成分较多,也可能提示红细胞裂解不充分、环境 RNA 污染或红细胞相关 reads 混入。对于 PBMC 数据来说,成熟红细胞通常没有细胞核,理论上不应大量出现在单细胞转录组中,因此高血红蛋白比例可能影响后续分析,需要特别关注。
# 标记线粒体基因:
# 人类线粒体基因通常以 "MT-" 开头;小鼠线粒体基因通常以 "mt-" 开头
adata.var["mt"] = adata.var_names.str.startswith("MT-") # 人类
# adata.var["mt"] = adata.var_names.str.startswith("mt-") # 小鼠
# 标记核糖体蛋白基因:
# 人类和小鼠中常见的核糖体蛋白基因通常以 "RPS"/"RPL" 或 "Rps"/"Rpl" 开头
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL")) # 人类
# adata.var["ribo"] = adata.var_names.str.startswith(("Rps", "Rpl")) # 小鼠
# 标记血红蛋白相关基因:
# 人类血红蛋白基因通常以 "HB" 开头;小鼠通常以 "Hb" 开头
adata.var["hb"] = adata.var_names.str.contains(r"^HB[ABDEGMQZ]\d*(?!\w)") # 人类
# adata.var["hb"] = adata.var_names.str.contains(r"^Hb[ab]") # 小鼠
计算细胞指标
sc.pp.calculate_qc_metrics(
adata, # 输入的 AnnData 对象,包含单细胞表达矩阵和注释信息
qc_vars=["mt", "ribo", "hb"], # 指定需要单独统计的基因集合:线粒体、核糖体蛋白和血红蛋白基因等关注的基因集
inplace=True, # 将计算结果直接写入 adata.obs 和 adata.var
percent_top=[20], # 计算每个细胞中表达量最高的前 20 个基因占总 counts 的比例
log1p=True# 同时计算 log(1+x) 转换后的 QC 指标
)
adata
AnnData object with n_obs × n_vars = 2700 × 32738
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb'
var: 'gene_ids', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
该函数向 .var 和 .obs 添加了几列新的信息。
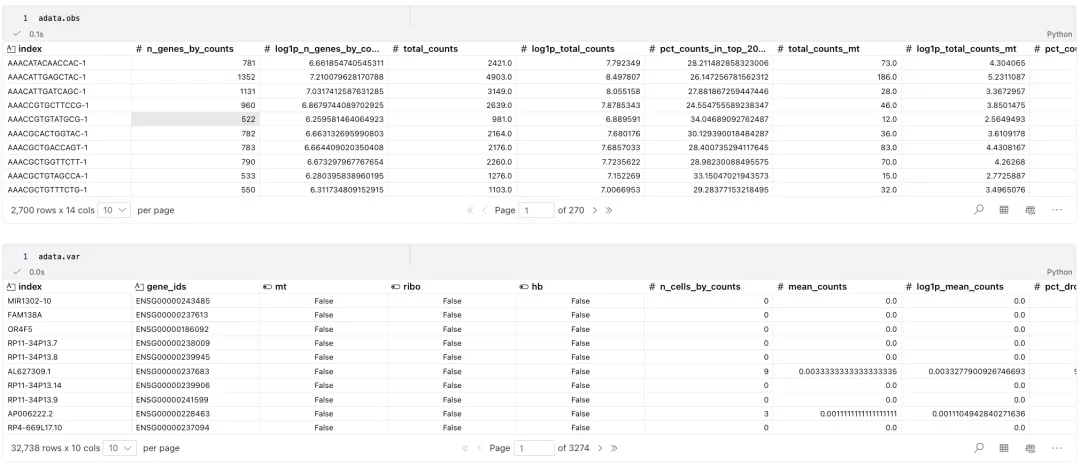
可视化细胞指标
我们经常关注以下几个指标:
n_genes_by_counts:每个细胞中检测到表达的基因数量,即 counts 大于 0 的基因数。total_counts:每个细胞的总 counts 数,也称为 library size。pct_counts_mt:每个细胞中线粒体基因 counts 占总 counts 的比例。
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
**{"color": "#f8766d"}, # 使用额外的参数来设置颜色
multi_panel=True
)
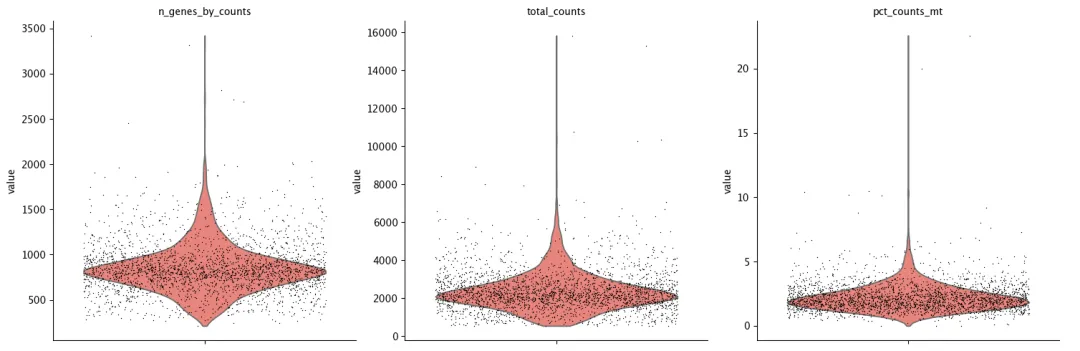
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
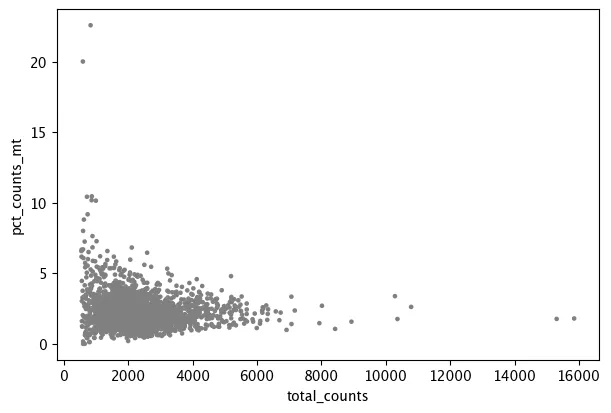
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")
根据指标过滤
在单细胞质控中,低质量细胞可通过两种方式进行过滤:一种是根据经验或数据分布手动设定阈值,例如过滤低 total_counts、低 n_genes_by_counts 或高 pct_counts_mt 的细胞;另一种是基于中位数绝对偏差(MAD)自动识别异常值,即将偏离整体中位数超过一定倍数 MAD 的细胞标记为异常。手动阈值直观、便于结合生物学背景判断,而 MAD 方法更适合大规模数据集,具有较好的稳健性和自动化程度。实际分析中可结合两种方法,并尽量采用相对宽松的过滤标准,以避免误删真实细胞群或稀有亚群。
手动过滤
基于上面的一些图表,现在可以手动设定一些过滤细胞的阈值,这一步并没有一个统一的标准。
adata = adata[adata.obs.n_genes_by_counts < 2000, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
adata.shape
(2634, 32738)
注意:如果你在一段代码中连续对数据进行多次筛选或其他操作,可以在最后一步使用 .copy() 来确保最终结果是一个独立的副本。这样可以避免在中间步骤中不小心修改原始数据。
基于中位数绝对偏差过滤
创建一个过滤函数
import numpy as np
from scipy.stats import median_abs_deviation
defis_outlier(adata, metric: str, nmads: int):
M = adata.obs[metric]
outlier = (M < np.median(M) - nmads * median_abs_deviation(M)) | (
np.median(M) + nmads * median_abs_deviation(M) < M
)
return outlier
根据考虑的指标设置过滤参数
adata.obs["outlier"] = (
is_outlier(adata, "log1p_total_counts", 5)
| is_outlier(adata, "log1p_n_genes_by_counts", 5)
| is_outlier(adata, "pct_counts_in_top_20_genes", 5)
)
adata.obs.outlier.value_counts()
adata.obs["mt_outlier"] = is_outlier(adata, "pct_counts_mt", 3) | (
adata.obs["pct_counts_mt"] > 8
)
adata.obs.mt_outlier.value_counts()
过滤
adata = adata[(~adata.obs.outlier) & (~adata.obs.mt_outlier)].copy()
过滤后可再次可视化查看过滤情况考虑是不是需要调整参数再次过滤。
碎碎念:因考虑写一套基于python全流程分析的单细胞推文,但很多过滤手段都是基于R的版本,此部分目前为基础标准流程,后续可能会有测试稳定的质控内容作为可选,特此说明和预告
参考书:《Single-cell best practices》
特别鸣谢:小洁老师

