前言
上一期使用R语言对GSE189357单细胞数据集进行了基础的分析,有同学留言有没有python相关的教程。近年来,随着单细胞测序技术的快速发展和测序成本的不断下降,越来越多的研究开始纳入大规模样本甚至百万级细胞的数据分析。在这种背景下,Python 生态中的 Scanpy、AnnData 和Omicverse等工具凭借其较高的计算效率及良好的扩展性,逐渐成为单细胞数据分析的重要选择。虽然目前 Seurat仍然是最受欢迎的单细胞分析工具之一,但对于大规模数据集Python也展现出了独特的优势。今天基于GSE189357数据集对Scanpy处理单细胞流程进行简单的整理。
我之前也是学习技能树曾老师他们的分析脚本,感兴趣的朋友也可以关注他们学习更多相关的知识。
数据选择
使用上期整理好的单细胞数据数据读入Python处理,没有整理的大家根据上期内容整理就行。单细胞转录组下游分析流程整理
数据读取
导入相应的python包
import scanpy as scimport numpy as npimport pandas as pdimport scipy.sparse as spsc.set_figure_params(figsize=(5, 5))
查看我们的数据文件夹
import osos.listdir('./GSE189357_RAW/')
批量读取数据
inputdir = "./data/raw/GSE189357_RAW"samples = sorted(os.listdir(inputdir))adata = {}for sample in samples: path = os.path.join(inputdir, sample) data = sc.read_10x_mtx( path, var_names="gene_symbols", cache=True ) data.var_names_make_unique() data.obs["sample"] = sample name = sample adata[name] = data print(name)
数据合并
adata = sc.concat(adata,label='sampleid')adata.obs_names_make_unique()adata
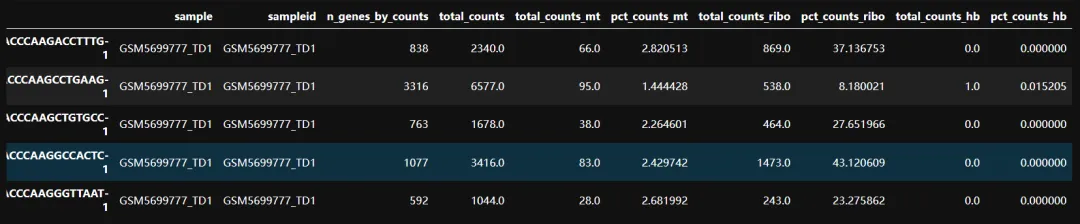
计算质控指标:线粒体基因比例;核糖体基因比例和红血细胞基因比例
# 计算线粒体基因比例adata.var['mt']=adata.var_names.str.startswith('MT-')# 计算核糖体基因比例adata.var['ribo']=adata.var_names.str.match('^RP[SL]')# 计算红血细胞基因比例adata.var['hb']=adata.var_names.str.match('^HB[^(P)]')# 计算sc.pp.calculate_qc_metrics(adata,qc_vars=['mt','ribo','hb'],percent_top=None,log1p=False,inplace=True)# 质控adata.obs.head()
这时候发现各个指标已经添加到了我们的obs当中。
可视化细胞的上述比例情况
from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (6, 4)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="sampleid", jitter=0.3,rotation=90,multi_panel=True, stripplot=False, # remove the internal dots inner="box" )
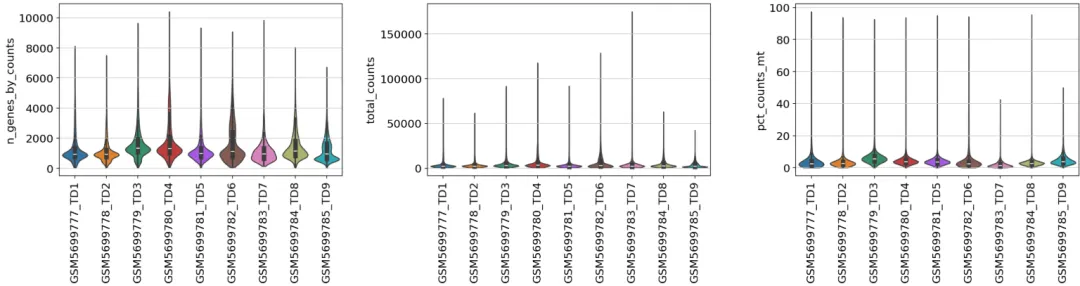
过滤细胞
# 基本过滤sc.pp.filter_genes(adata, min_cells=3)## 过滤# 方法2:一次性过滤所有条件(更高效)adata = adata[ (adata.obs['n_genes_by_counts'] > 300) & (adata.obs['n_genes_by_counts'] < 5000) & (adata.obs['total_counts'] > 250) & # (adata.obs['total_counts'] < 3000000) & (adata.obs['pct_counts_mt'] < 20), :].copy()## 过滤后的数据adata.shape
质控后再看看
from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (6, 4)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="sampleid", jitter=0.4,rotation=90,multi_panel=True, stripplot=False, inner="box" )
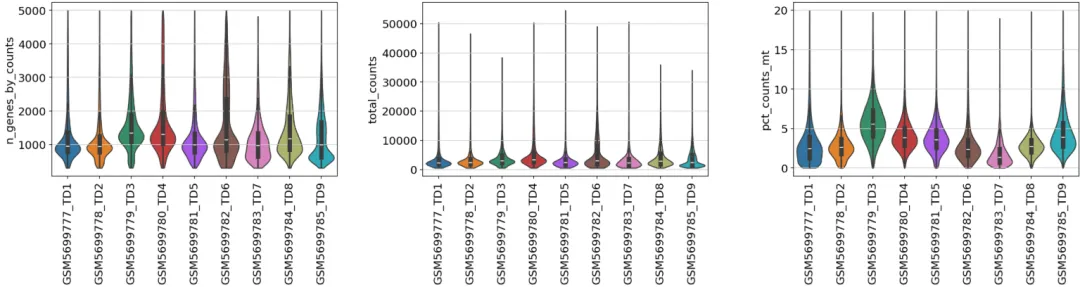
降维聚类
保存数据
adata.write('./data/processed/adata_raw.h5ad')
这里去批次效应还是用harmony
# 首先将数据矩阵标准化(校正文库大小):adata.layers["counts"] = adata.X.copy()adata.raw = adata.copy()sc.pp.normalize_total(adata,target_sum=1e4)# 对数据进行logsc.pp.log1p(adata)# 高变基因sc.pp.highly_variable_genes(adata)# 归一化sc.pp.scale(adata)# pca分析sc.tl.pca(adata,use_highly_variable=True)# harmony整合#--------------------------------------------------------------------# sc.external.pp.harmony_integrate(adata,'sampleid') # 这里用自己的样本名就行,我的是sampleid## 我发现我运行这个函数会报错,原因是sc.external.pp.harmony_integrate运行完的数据不符合adata.obsm中的数据储存形式,正好是反过来的,可能是软件版本问题,所以用了以下方法#--------------------------------------------------------------------import harmonypy as hmho = hm.run_harmony( adata.obsm["X_pca"], adata.obs,"sample")adata.obsm["X_pca_harmony"] = ho.Z_corr#--------------------------------------------------------------------# 聚类sc.pp.neighbors(adata,use_rep='X_pca_harmony',n_pcs=50)# sc.pp.neighbors(adata,use_rep='X_pca',n_pcs=30)sc.tl.umap(adata)sc.tl.tsne(adata)# Using the igraph implementation and a fixed number of iterations can be significantly faster, especially for larger datasetssc.tl.leiden(adata, flavor="igraph", n_iterations=2,resolution=0.5)
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (6.5, 6)}): sc.pl.umap(adata, color=["leiden"], legend_loc="on data", size=7)
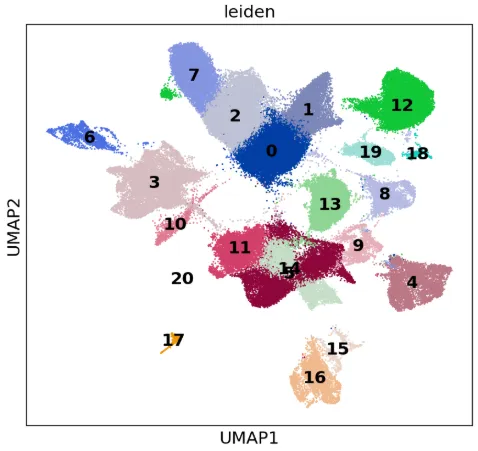
with rc_context({"figure.figsize": (6.5, 6)}): sc.pl.umap(adata, color=['sample'],size=7)
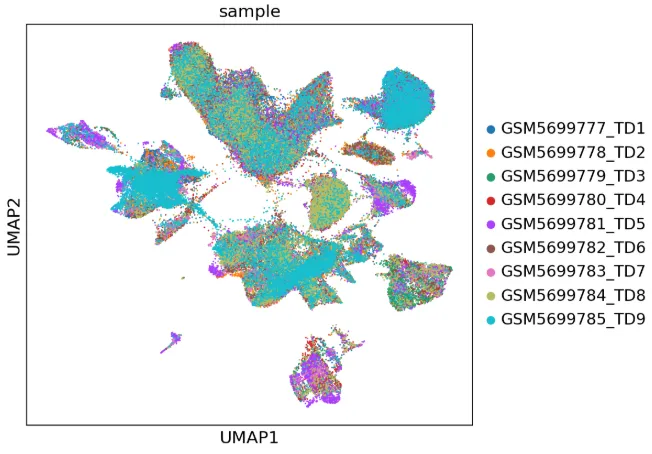
细胞注释
marker = {"T cell": ["CD3D", "CD3E"],"NK": ["NKG7", "GNLY"],"B cells": ["CD79A", "CD79B", "CD19"],"Plasma": ["MZB1", "IGHG1"],"Myeloid": ["LYZ", "TPSB2", "AIF1", "HLA-DRA", "HLA-DRB1", "CPA3"],"Epithelial": ["EPCAM", "SCGB1A1"],"Endothelial": ["CLDN5", "PECAM1"]}sc.pl.dotplot(adata, marker, groupby="leiden", standard_scale="var", show =False# save = '2.png' )
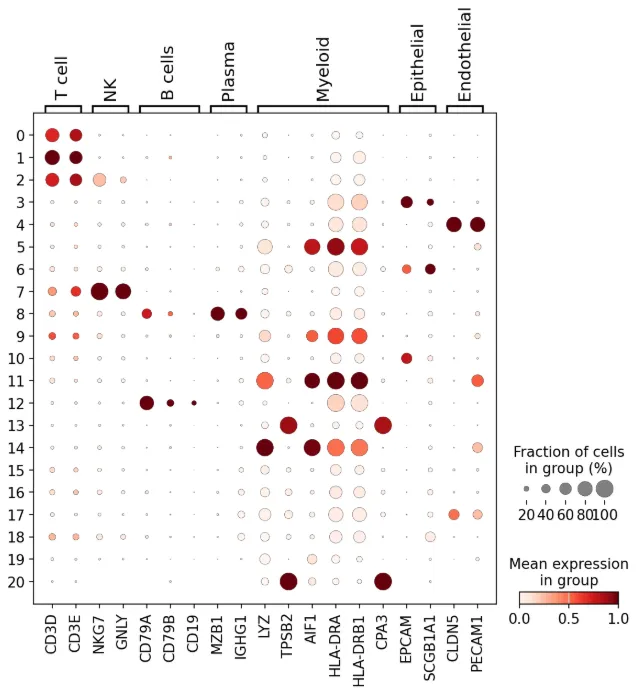
这里我注释的比较草率,大家自己的数据要认真注释
adata.obs["celltype"] = "unknown"adata.obs.loc[adata.obs["leiden"].isin(["0", "1","2","7"]), "celltype"] = "T&NK"adata.obs.loc[adata.obs["leiden"].isin(["12"]), "celltype"] = "B cell"adata.obs.loc[adata.obs["leiden"].isin(["8"]), "celltype"] = "Plasma"adata.obs.loc[adata.obs["leiden"].isin(["5", "9", "10", "11", "13", "14", "15","16","18","19","20"]), "celltype"] = "Myeloid"adata.obs.loc[adata.obs["leiden"].isin(["3", "6", "10"]), "celltype"] = "Epithelial"adata.obs.loc[adata.obs["leiden"].isin(["4", "17"]), "celltype"] = "Endothelial"

按细胞类型画一下marker基因的dotplot
marker = {"T cell": ["CD3D", "CD3E"],"NK": ["NKG7", "GNLY"],"B cells": ["CD79A", "CD79B", "CD19"],"Plasma": ["MZB1", "IGHG1"],"Myeloid": ["LYZ", "TPSB2", "AIF1", "HLA-DRA", "HLA-DRB1", "CPA3"],"Epithelial": ["EPCAM", "SCGB1A1"],"Endothelial": ["CLDN5", "PECAM1"]}sc.pl.dotplot(adata, marker, groupby="celltype", standard_scale="var", show =False# save = '2.png' )
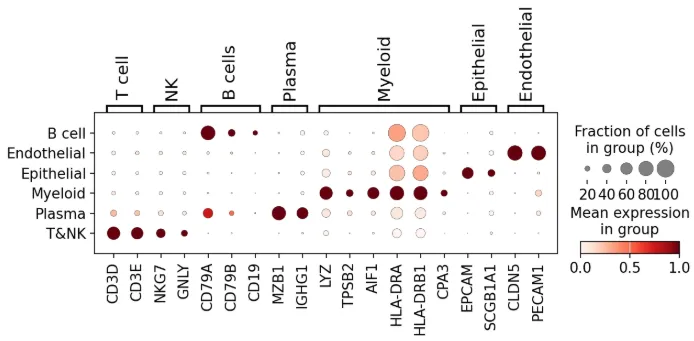
保存数据
adata.write('./data/processed/adata_anno.h5ad')
画细胞比例图
import matplotlib.pyplot as pltfrom matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (7, 5)}): ax = pd.crosstab( adata.obs['sample'], adata.obs['celltype'], normalize='index' ).plot( kind='bar', stacked=True, xlabel='' ) plt.legend( bbox_to_anchor=(1.05, 1), loc='upper left', frameon=False ) plt.tight_layout()
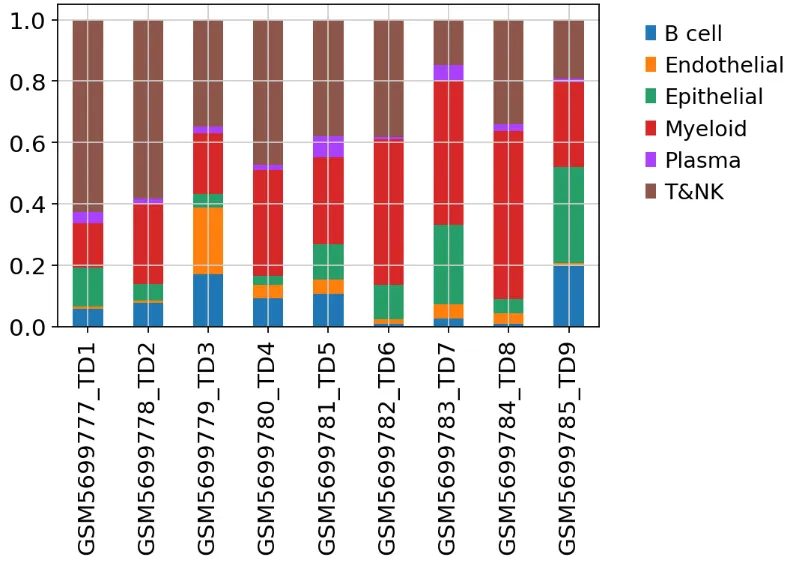
写在最后
本次内容分享了scanpy是最基础的分析,对于单细胞的一些高级分析后续再分享,欢迎大家持续关注与交流。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
