python单细胞流程-数据整合
- 2026-07-02 16:31:26
为什么要进行数据整合?
在单样本流程中,数据通常来自同一个 library。经过质控、归一化、高变基因选择、降维、聚类和注释后,可以得到一个相对完整的分析结果。
真实项目中更常见的情况是多个样本共同分析。例如:
样本 1:control / donor1 / batch1
样本 2:control / donor2 / batch1
样本 3:disease / donor3 / batch2
样本 4:disease / donor4 / batch2
如果直接合并所有细胞后做 PCA 和 UMAP,细胞之间的差异可能同时包含两部分信息:
真实生物学差异:细胞类型、细胞状态、疾病或处理效应。 技术差异:建库批次、测序深度、样本处理、10X chemistry 或测序 lane。
数据整合(data integration)或批次校正(batch correction)的目的,是在共同分析多个样本时减弱不希望保留的技术差异,同时尽量保留真实的生物学结构。
需要注意的是,整合不是为了把所有样本强行混在一起。好的整合结果应该是:
同一细胞类型内部,不同样本适度混合
不同细胞类型或真实状态之间,仍然清楚分开
如果整合后 UMAP 很漂亮,但 marker gene 变得模糊、疾病特异状态消失、稀有细胞群被合并掉,反而可能是过度整合。
数据整合在流程中的位置
单样本基础流程是:
-> 数据清洗与质控
-> 数据归一化
-> 高变基因选择
-> 降维
-> 聚类
-> 注释
多样本项目中,需要先把每个样本读入并完成基础质控,再合并到同一个 AnnData 对象。整合通常发生在构建最终邻居图和聚类之前:
-> 每个样本单独读取
-> 每个样本基础 QC
-> 合并 AnnData
-> 数据归一化
-> 高变基因选择
-> PCA
-> 数据整合
-> 基于整合后的表示构建邻居图
-> UMAP / Leiden 聚类
-> marker gene 和细胞类型注释
其中,“合并 AnnData”是多样本分析的准备步骤。它只是把多个样本的细胞按行拼接到同一个 adata 中,同时在 adata.obs 中保留每个细胞对应的 sample、donor、batch 和 condition 信息。
PCA 本身只是在高变基因表达矩阵上得到一个初始低维空间,并不会自动去除批次效应。如果批次效应很强,前几个 PC 可能主要反映样本或批次差异。Harmony 会在 PCA 空间中进一步校正批次;scVI 则使用原始 counts 和 batch 信息学习新的 latent representation。
整合后的低维表示主要用于:
构建邻居图。 UMAP 可视化。 Leiden 聚类。 共同细胞结构识别。 参考映射或 label transfer。
条件间差异表达、pseudobulk 或严格的统计比较不应直接使用整合后的 embedding。更稳妥的做法是保留原始 counts,并在后续使用 pseudobulk 或能处理样本重复的统计模型。
整合表示:用于找结构
原始 counts:用于做统计比较
本节示例数据
数据集:
https://www.10xgenomics.com/datasets/60k_Human_TotalSeqB_hashing_3p_gem-x
60k Human PBMCs Stained with 8 TotalSeq-B Human Hashtags
Donor 1-4, 4 samples
数据结构:
物种:human。 样本:4 个 PBMC donor。 疾病状态:4 个 donor 均为 diseased human donors。 技术:Chromium GEM-X Single Cell 3'。 Feature Barcode:TotalSeq-B human hashtag。 输入矩阵:Cell Ranger count输出的 per-sample filtered feature-barcode HDF5。
每个 donor 下载同一类文件:
Gene Expression - Feature / cell matrix HDF5 (per-sample)
下载完成后,本节需要 4 个 .h5 文件:
Donor1: *_Human_Donor1_*_count_sample_filtered_feature_bc_matrix.h5
Donor2: *_Human_Donor2_*_count_sample_filtered_feature_bc_matrix.h5
Donor3: *_Human_Donor3_*_count_sample_filtered_feature_bc_matrix.h5
Donor4: *_Human_Donor4_*_count_sample_filtered_feature_bc_matrix.h5
这里使用的是已经拆分好的 per-sample 矩阵,不再从 raw multiplex matrix 重新做 hashtag demultiplexing。TotalSeq-B hashtag 用于样本标记,不等同于 TBNK antibody cocktail 这类细胞表面蛋白 panel。
加载库
import os
import numpy as np
import pandas as pd
import scanpy as sc
import anndata as ad
import harmonypy as hm # Harmony
准备样本信息表
多样本分析中,样本信息应单独整理成表格,并写入 adata.obs。后续整合、分组统计和结果解释都依赖这些字段。
project_dir = "/home/data/t090639/project/pbmc_60k"
data_dir = f"{project_dir}/data"
processed_dir = f"{project_dir}/processed"
os.makedirs(processed_dir, exist_ok=True)
建立样本信息表:
sample_metadata = pd.DataFrame({
"sample": ["Donor1", "Donor2", "Donor3", "Donor4"],
"path": [
f"{data_dir}/60k_Human_TotalSeqB_hashing_3p_gem-x_Human_Donor1_PBMC_TotalSeqB_hashing_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5",
f"{data_dir}/60k_Human_TotalSeqB_hashing_3p_gem-x_Human_Donor2_PBMC_TotalSeqB_hashing_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5",
f"{data_dir}/60k_Human_TotalSeqB_hashing_3p_gem-x_Human_Donor3_PBMC_TotalSeqB_hashing_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5",
f"{data_dir}/60k_Human_TotalSeqB_hashing_3p_gem-x_Human_Donor4_PBMC_TotalSeqB_hashing_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5",
],
"donor": ["Donor1", "Donor2", "Donor3", "Donor4"],
"condition": ["diseased", "diseased", "diseased", "diseased"],
"batch": ["Donor1", "Donor2", "Donor3", "Donor4"],
"chemistry": ["GEM-X_3p", "GEM-X_3p", "GEM-X_3p", "GEM-X_3p"],
"feature_barcode": ["TotalSeq-B_hashtag"] * 4,
})
sample_metadata
检查文件路径:
sample_metadata["path"]
读取每个样本
本节只使用 Gene Expression 矩阵做 RNA 数据整合。10X HDF5 文件可以直接用 sc.read_10x_h5() 读取;gex_only=True 表示只保留 Gene Expression feature。
如果设置 gex_only=False,同一个 feature-barcode HDF5 中的其他 feature type 也会一起读入,例如 Antibody Capture、CRISPR Guide Capture 或 Custom。本数据集的 TotalSeq-B hashtag 属于 Feature Barcode 信息,但本节只做 RNA 整合,因此保持 gex_only=True。
metadata_cols = ["sample", "donor", "condition", "batch", "chemistry", "feature_barcode"]
adata_list = []
for _, row in sample_metadata.iterrows():
adata_i = sc.read_10x_h5(row["path"], gex_only=True)
adata_i.var_names_make_unique()
for col in metadata_cols:
adata_i.obs[col] = row[col]
adata_i.obs_names = [
f"{row['sample']}_{barcode}"
for barcode in adata_i.obs_names
]
adata_i.layers["counts"] = adata_i.X.copy()
adata_list.append(adata_i)
如果读入时提示 Variable names are not unique,通常是因为少数基因 symbol 重名。这里的 adata_i.var_names_make_unique() 会在基因名后自动添加后缀,使其变成唯一名称,不影响后续分析。
查看每个对象:
for adata_i in adata_list:
print(
adata_i.obs["sample"].iloc[0],
adata_i.shape
)
如果后续需要分析 hashtag feature 或 ADT protein feature,可以把 gex_only 改成 False,再单独提取非 Gene Expression feature。本节只做 RNA 数据整合,因此保持 gex_only=True。
每个样本基础 QC
多样本数据应先在每个样本内部做基础 QC,再合并。这样可以避免某个低质量样本在合并后被整体分布掩盖。
计算每个样本的 QC 指标:
for adata_i in adata_list:
adata_i.var["mt"] = adata_i.var_names.str.startswith("MT-")
adata_i.var["ribo"] = adata_i.var_names.str.startswith(("RPS", "RPL"))
adata_i.var["hb"] = adata_i.var_names.str.contains(r"^HB[ABDEGMQZ]\d*(?!\w)")
sc.pp.calculate_qc_metrics(
adata_i,
qc_vars=["mt", "ribo", "hb"],
percent_top=[20],
log1p=True,
inplace=True
)
整理每个样本的 QC 概览:
qc_cols = [
"total_counts",
"n_genes_by_counts",
"pct_counts_mt",
"pct_counts_ribo",
"pct_counts_hb",
]
qc_summary = pd.concat([
adata_i.obs[qc_cols]
.assign(sample=adata_i.obs["sample"].iloc[0])
.groupby("sample", observed=True)
.median()
for adata_i in adata_list
])
qc_summary
可视化每个样本的基础 QC 分布:
for adata_i in adata_list:
print(adata_i.obs["sample"].iloc[0])
sc.pl.violin(
adata_i,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True
)
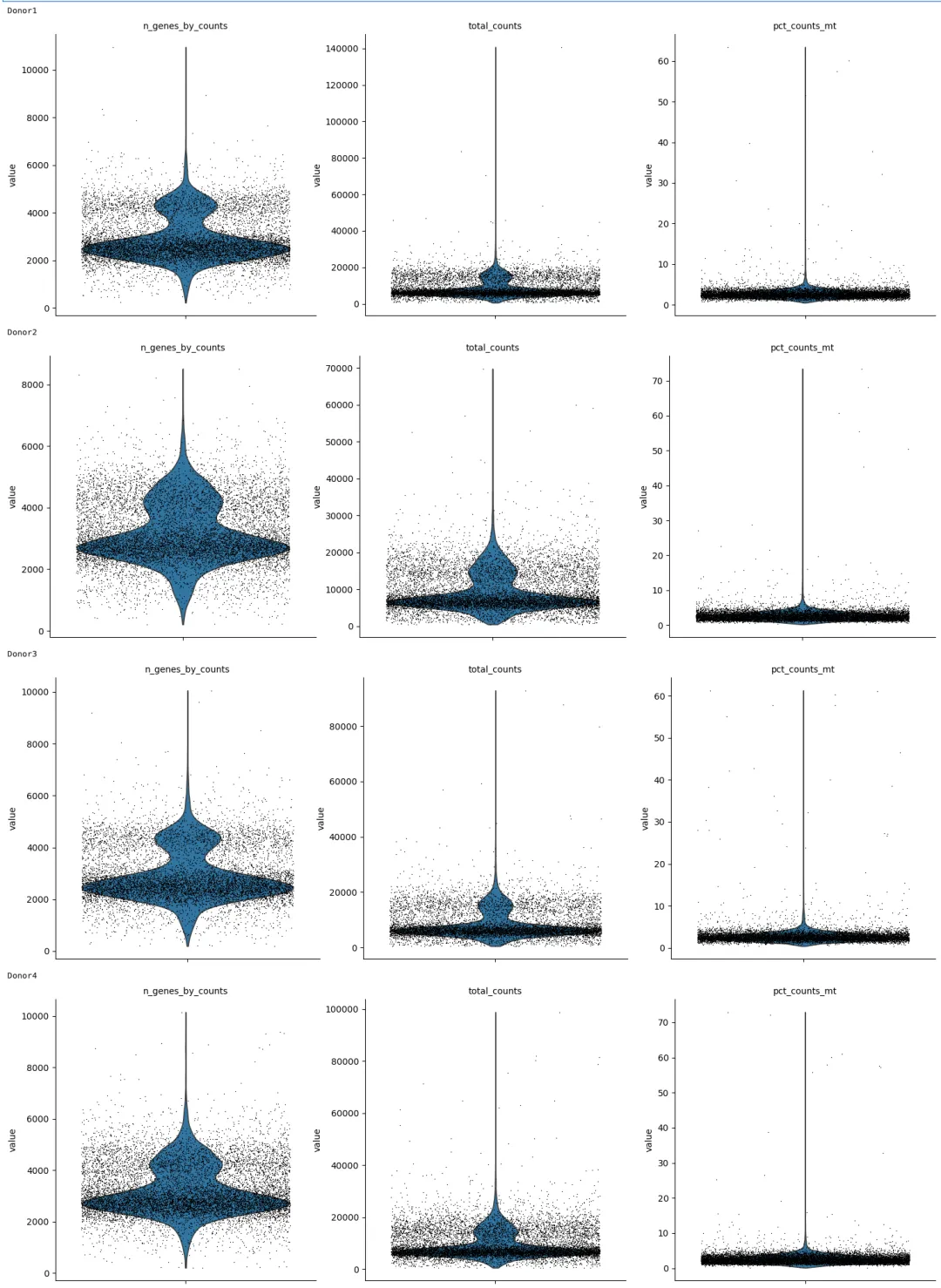
根据 PBMC 数据的常见分布,先使用相对宽松的过滤标准:
min_genes = 200
max_genes = 6000
max_mt = 15
filtered_list = []
for adata_i in adata_list:
adata_i = adata_i[
(adata_i.obs["n_genes_by_counts"] >= min_genes)
& (adata_i.obs["n_genes_by_counts"] <= max_genes)
& (adata_i.obs["pct_counts_mt"] < max_mt)
].copy()
filtered_list.append(adata_i)
adata_list = filtered_list
这里的阈值只是 PBMC 示例数据的起点。实际项目中,应先看每个样本自己的 QC 分布,再决定过滤标准。对于肿瘤、组织样本、冷冻样本或 snRNA-seq,pct_counts_mt 和 n_genes_by_counts 的阈值都不应直接照搬。
合并 AnnData 对象
完成单样本基础 QC 后,将 4 个样本合并到同一个 AnnData 对象中。
adata = ad.concat(
adata_list,
join="outer",
index_unique=None,
merge="same"
)
adata

检查合并结果:
print(adata.shape)
print(adata.obs_names.is_unique)
adata.obs[["sample", "donor", "condition", "batch", "chemistry"]].head()

查看每个样本保留下来的细胞数:
adata.obs["sample"].value_counts().sort_index()
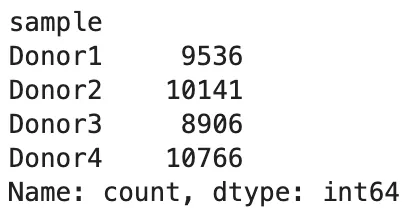
查看样本和分组关系:
pd.crosstab(adata.obs["sample"], adata.obs["condition"])
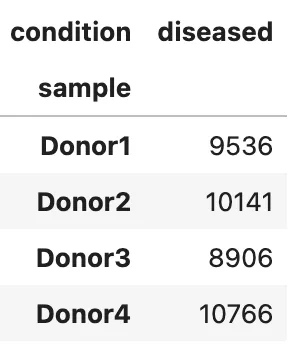
本示例的 4 个 donor 都来自 diseased human donors,因此不用于演示病例/对照差异分析。实际病例/对照项目中,应特别检查 batch 和 condition 是否完全混淆。
pd.crosstab(adata.obs["batch"], adata.obs["condition"])
如果出现某个 batch 全部是 control、另一个 batch 全部是 disease,说明技术批次和研究分组完全重叠。此时整合可以用于探索共同细胞类型,但不能可靠地区分技术差异和疾病差异。
保存合并后的对象:
merged_out = f"{processed_dir}/08_pbmc_60k_merged_qc.h5ad"
adata.write_h5ad(merged_out)
归一化和高变基因选择
读取合并后的对象:
adata = sc.read_h5ad(merged_out)
adata

执行 Log(CP10k+1) 归一化:
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
保存完整的 Log(CP10k+1) 表达矩阵:
adata.raw = adata.copy()
多样本数据做高变基因选择时,建议指定 batch_key="batch"。这样可以优先选择在多个样本中稳定变异的基因,减少只由单个样本驱动的高变基因。
sc.pp.highly_variable_genes(
adata,
n_top_genes=3000,
flavor="seurat",
batch_key="batch"
)
查看高变基因结果:
adata.var["highly_variable"].value_counts()
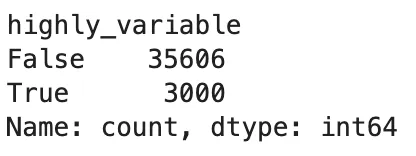
sc.pl.highly_variable_genes(adata)
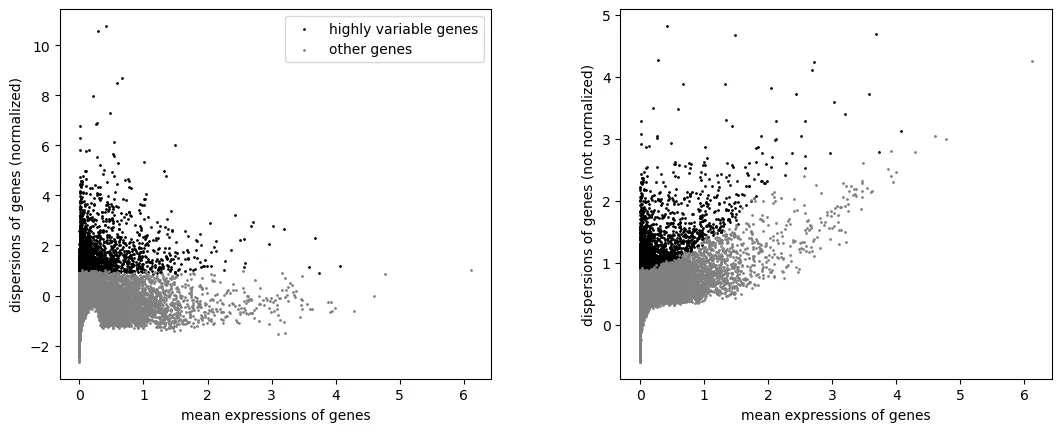
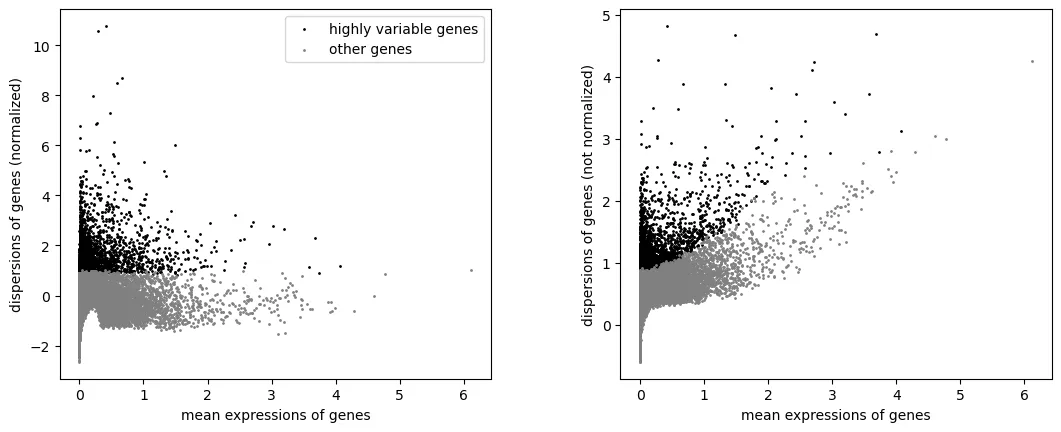
只保留高变基因用于 PCA 和整合:
adata = adata[:, adata.var["highly_variable"]].copy()
adata

这里不会影响 adata.raw。后续 marker gene 可视化仍然可以从 adata.raw 中读取完整的 Log(CP10k+1) 表达矩阵。
保存:
hvg_out = f"{processed_dir}/09_pbmc_60k_normalized_hvg.h5ad"
adata.write_h5ad(hvg_out)
PCA 和未整合结果
数据整合前,应先建立一个未整合 baseline。这样后面才能判断整合是否真正改善了 batch effect。
adata = sc.read_h5ad(hvg_out)
adata
Scaling:
sc.pp.scale(adata, max_value=10)
PCA:
sc.pp.pca(
adata,
n_comps=50,
svd_solver="arpack",
random_state=0
)
查看 PCA 是否被样本或 QC 指标驱动:
plot_cols = [
"total_counts",
"n_genes_by_counts",
"pct_counts_mt",
"sample",
"batch",
"condition",
]
sc.pl.pca(
adata,
color=plot_cols,
ncols=3
)
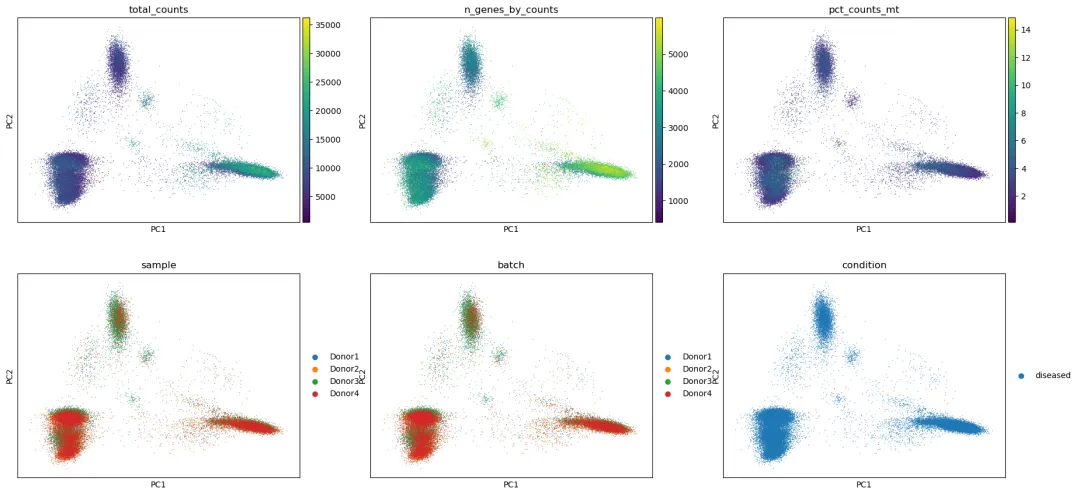
选择 PC 数:
sc.pl.pca_variance_ratio(adata, n_pcs=50, log=True)
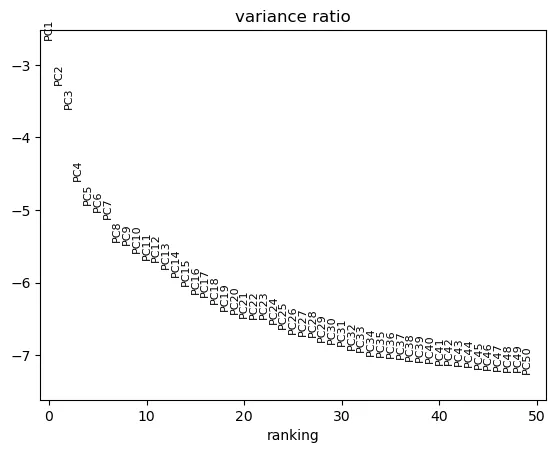
n_pcs = 30
构建未整合邻居图:
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=n_pcs,
random_state=0
)
运行 UMAP:
sc.tl.umap(
adata,
min_dist=0.3,
spread=1.0,
random_state=0
)
运行 Leiden 聚类:
sc.tl.leiden(
adata,
resolution=0.5,
key_added="leiden_res0_5",
flavor="igraph",
n_iterations=2,
random_state=0
)
查看未整合结果:
sc.pl.umap(
adata,
color=["sample", "batch", "condition", "leiden_res0_5"],
ncols=2
)
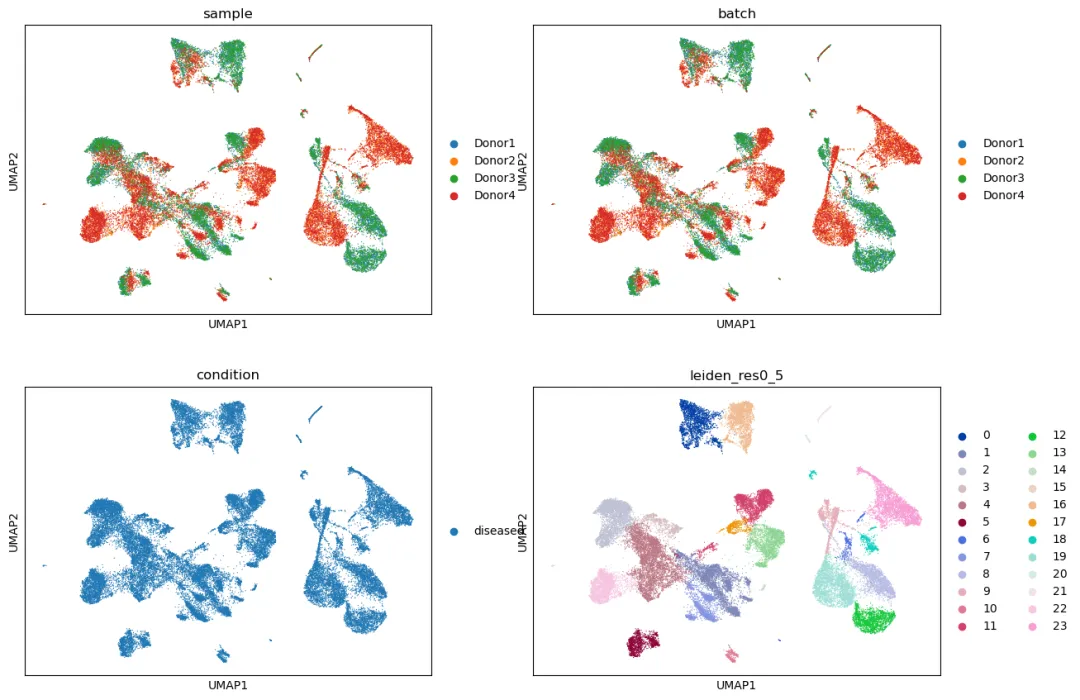
如果 UMAP 主要按 sample 或 batch 分开,而不是按细胞类型 marker 分开,说明需要进一步整合。
查看 PBMC 常见 marker:
marker_genes = ["MS4A1", "CD3D", "CD8A", "LYZ", "NKG7", "PPBP"]
sc.pl.umap(
adata,
color=marker_genes,
use_raw=True,
ncols=3,
vmax="p99"
)
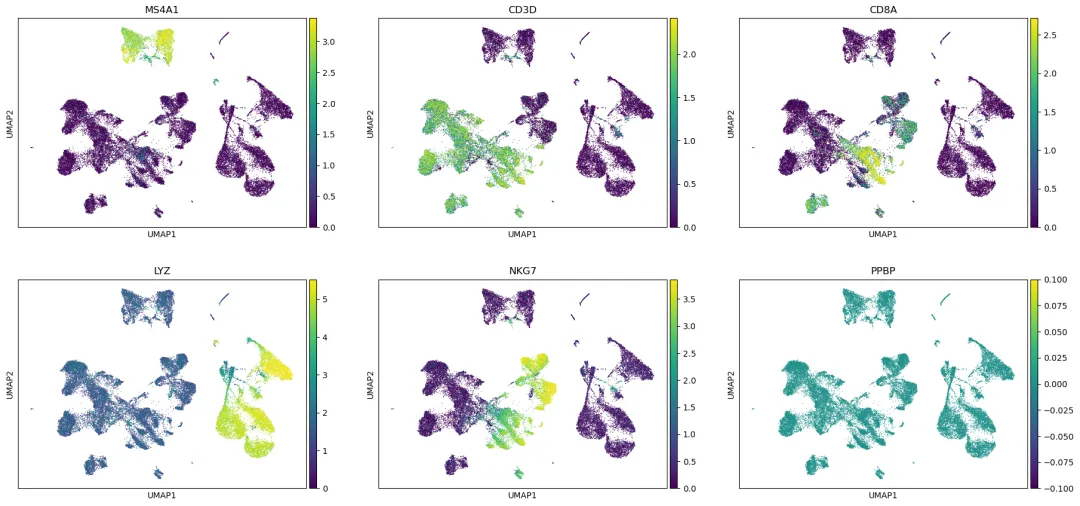
保存未整合 baseline:
baseline_out = f"{processed_dir}/10_pbmc_60k_preintegration_baseline.h5ad"
adata.write_h5ad(baseline_out)
Harmony 整合
Harmony 在 PCA 空间中校正 batch effect,不直接修改表达矩阵。
adata_harmony = sc.read_h5ad(baseline_out)
运行 Harmony:
harmony_out = hm.run_harmony(
adata_harmony.obsm["X_pca"],
adata_harmony.obs,
"batch",
random_state=0
)
adata_harmony.obsm["X_pca_harmony"] = np.asarray(harmony_out.Z_corr)
如果需要同时校正多个技术变量,可以把 key 写成列表:
# harmony_keys = ["batch", "chemistry"]
# harmony_out = hm.run_harmony(
# adata_harmony.obsm["X_pca"],
# adata_harmony.obs,
# harmony_keys,
# random_state=0
# )
# adata_harmony.obsm["X_pca_harmony"] = np.asarray(harmony_out.Z_corr)
不要把研究分组直接放进 harmony_keys。例如病例/对照、处理组/对照组、时间点等通常是后续要解释的生物学变量,不应作为 Harmony 要去除的 batch。
Harmony 的结果保存在:
adata_harmony.obsm["X_pca_harmony"].shape
基于 Harmony embedding 重新构建邻居图:
sc.pp.neighbors(
adata_harmony,
n_neighbors=15,
use_rep="X_pca_harmony",
random_state=0
)
重新运行 UMAP:
sc.tl.umap(
adata_harmony,
min_dist=0.3,
spread=1.0,
random_state=0
)
重新运行 Leiden:
sc.tl.leiden(
adata_harmony,
resolution=0.5,
key_added="harmony_leiden_res0_5",
flavor="igraph",
n_iterations=2,
random_state=0
)
查看 Harmony 整合结果:
sc.pl.umap(
adata_harmony,
color=["sample", "batch", "condition", "harmony_leiden_res0_5"],
ncols=2
)
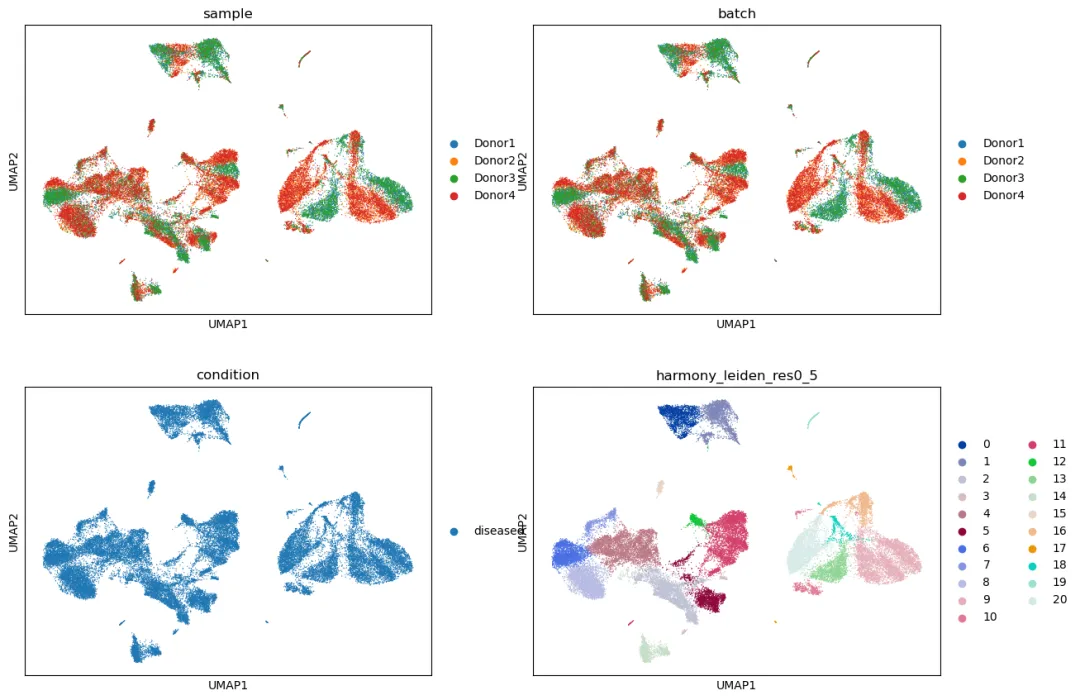
查看 marker gene 是否仍然清楚:
marker_genes = ["MS4A1", "CD3D", "CD8A", "LYZ", "NKG7", "PPBP"]
sc.pl.umap(
adata_harmony,
color=marker_genes,
use_raw=True,
ncols=3,
vmax="p99"
)
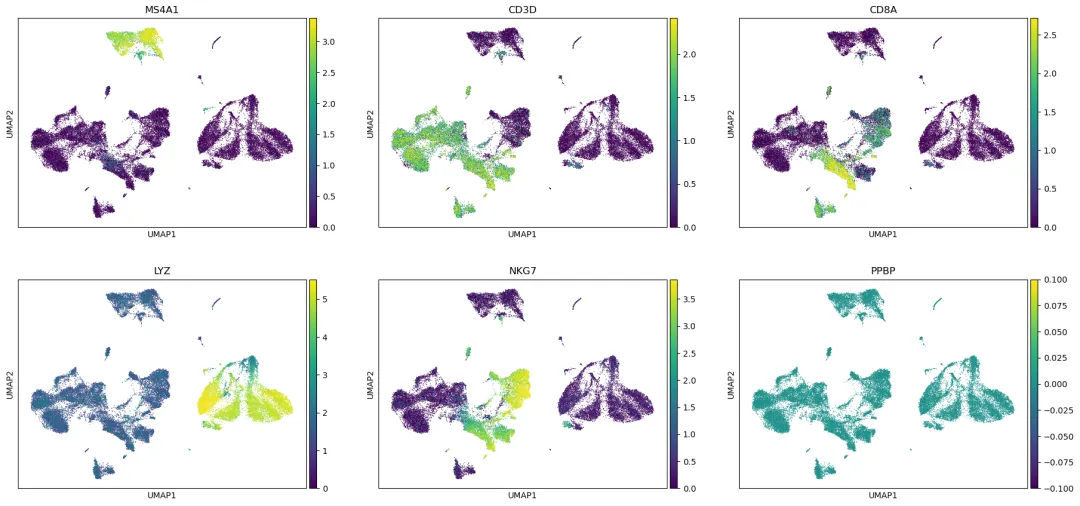
保存 Harmony 结果:
adata_harmony.uns["integration_method"] = "Harmony"
adata_harmony.uns["integration_representation"] = "X_pca_harmony"
adata_harmony.uns["integration_batch_key"] = "batch"
harmony_out = f"{processed_dir}/11_pbmc_60k_harmony_integrated.h5ad"
adata_harmony.write_h5ad(harmony_out)
Harmony 适合大多数同平台多样本项目。它速度快、结果直观,并且不会覆盖原始表达矩阵。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 运维还在死记 Linux 命令?这份大全存手机,排错 30 秒搞定
- 基于深度学习的蘑菇识别系统~Python+人工智能+模型训练+CNN算法
- 学Python能做什么?薪资多少?
- Python安装 (图文超级详细)
- Python编程班即将开课
- 100道Python真题(附答案),新手刷完直接弯道超车
- 苏州园区python培训——2026年Python彻底变了!告别“慢语言”标签,AI时代全员刚需很多人一直对有个刻板印象:简单好用
- Linux 根目录功能全览,一图秒懂!
- 基于 Linux 做商业系统,哪些代码必须开源,哪些可以闭源?
- 写给自己之php反序列化-快递打包员的故事
