从零开始的万字超详细Python单细胞转录组分析入门教程 (上):环境配置与Anndata基础
- 2026-06-30 15:15:19

写给考研复试的你:别慌,喵喵师兄带你从零开始!
写在前面
嗨,各位小伙伴们好呀!
因为最近遇到很多希望入门学习单细胞转录组的朋友,尤其是在考研复试前希望掌握一点有关科研的技术知识。
生信确实是一个比较快速能出结果并且不需要太多外部资源的东西,并且在目前的科研领域有较多的应用,许多课题组确实会偏向于在已有的湿实验之外加入一些干实验来丰富文章的内容,对于复试而言有这个技巧还是很加分的。
因此,本文目的就是在于帮你快速打好单细胞分析的地基,并且提供一些后续的学习资料。
本教程的目标
让你理解单细胞分析是怎么回事 让你的Windows/linux电脑跑起来Python单细胞分析环境(mac应该也有类似的方法,可以搜以下配置的教程,后面的代码应该都类似,就是我穷穷买不起苹果电脑(bushi)) 让你在复试时能自信地说:"单细胞分析?我了解一些!还针对导师的课题进行了一些公共数据分析,请老师看看我的结果!"
注意事项
在学习前有几个关键地方需要注意:
1.所谓"入门"指的是明白代码和IDE等这些基本的工具是什么东西,代码在哪里跑?文件怎么读取?结果会怎么输出?数据格式是什么? 在我看来这些才是阻挡初学者继续学习下去的主要问题。尽量先把这些搞清楚再去走下一步。 2.生信最好的学习资源永远是网络和github上的原版教程,在入门后最好去到这些python库或者R包的主页完整的学习,并搞清楚每个代码什么意思。大致了解流程后就可以去看一些高分的单细胞文章的代码写法,会对你的代码水平和有很大提升。 3.多用ai帮你理解代码和解决问题,但一定要理解ai做每一步的原因,不然等于什么都没学到,在以后还可能被ai带跑,导致项目出现重大错误。
第一章:为什么用Python做单细胞分析?
1.1 单细胞转录组测序是什么?
- Bulk RNA-seq
:测的是一堆细胞的平均基因表达 - scRNA-seq
:测的是每一个细胞的基因表达
这有什么用呢?想象一下,肿瘤组织里有各种各样的细胞,有的是癌细胞,有的是免疫细胞,有的是成纤维细胞。用bulk测序,你只能看到一个"混合信号";用单细胞测序,你能精确地看到每种细胞在干什么,甚至能发现那些数量稀少但很关键的细胞群。并且能具体探究某一类细胞中的变化。
1.2 为什么选Python而不是R?
说实话,做单细胞分析,R语言的Seurat包确实很强大,用的人也很多。但Python同样是一个绝佳的选择,原因如下:
python对电脑内存的需求没有R那么高,并且官方教程对结构的说明也比较清晰。了解完Anndata的结构之后,去了解Seurat V5的结构会非常迅速。 同时目前有较为成熟且快速的pipeline(虽然R也有SCOP包),比如我很喜欢的omicverse(重点推荐,除了环境配置比较麻烦外没有什么缺点)等,可以大大加速你跑各种复杂分析的效率,降低学习成本。 python调用GPU资源比较方便,可以大大加速运行效率,把你打游戏用的4090用起来。 后期肯定是R和python都要学的,数据在两个不同的语言里是可以互相转化的,你需要哪个工具做分析你就切换到对应的语言和环境去做。
喵喵师兄的建议:如果你是编程零基础,或者未来想往机器学习/深度学习方向发展,强烈推荐从Python入手!
1.3 第一步:认识你的命令行工具 CMD/PowerShell/WSL/linux
在开始之前,我们需要认识电脑的命令行界面。这是后续所有操作的基础
什么是命令行?
命令行就是你跟电脑"对话"的一种方式。平时我们用鼠标点点点,其实本质上都是在执行命令。命令行就是直接用键盘输入命令,让电脑干活。
Windows上的命令行工具:
- CMD(命令提示符)
Windows自带的老古董 按 Win + R,输入cmd,回车就能打开功能比较基础,但日常够用(我平常基本上都用的cmd) - PowerShell
CMD的升级版,功能更强大 Windows 10/11默认的命令行工具 右键开始菜单就能找到 - WSL(Windows Subsystem for Linux)/ Linux
让你在Windows里运行Linux系统 很多生信工具在Linux下运行更顺畅
为什么要用WSL?
虽然我们这个教程主要使用原生Windows环境就够了,但了解WSL很重要:
很多生物信息学工具原生支持Linux 以后接触生信的上游数据和高通量测序数据处理,WSL会是你的好帮手 面试时说"我会用linux",绝对加分。 重要的python单细胞库omicverse在linux环境的安装更为方便。
1.4 在Windows上安装WSL(可选但推荐)
注意:如果你只是想快速入门单细胞分析,可以暂时跳过安装wsl。但如果时间充裕,强烈建议装上!会让你在后续节省很多时间。
安装步骤(超级简单):
第一步:打开PowerShell(管理员模式)
在开始菜单中搜索powershell 选择"Windows PowerShell(管理员)"或"终端(管理员)"
或者直接按Win + R,输入cmd,回车,用cmd也行
第二步:一行命令搞定安装
复制粘贴进去,按回车
wsl --install -d Ubuntu-22.04
就这一行!Windows会自动帮你:
启用WSL功能 下载并安装Ubuntu(默认的Linux发行版)
第三步:重启电脑
重启电脑,打开cmd输入wsl,系统会提示为ubuntu创建用户名和密码(随便设个简单点的就行)。
第四步:验证安装
wsl --version
小贴士:
用户名一定要用英文,别用中文 密码输入时不会显示,别以为键盘坏了 以后想进入Linux,打开cmd输入wsl,就会进入虚拟机了。
这里需要记住一些linux的基础常用命令:
#linux(特指wsl)的目录指示格式,如假设在你D盘里的miaomiao文件夹中的miaomiao.md文件像这样表示
/mnt/d/miaomiao/miaomiao.md
#进入目录(文件夹)
cd XX文件夹
#回到上级目录
cd ..
#查看目录结构(你现在在的文件夹里有哪些文件)
ls
#创建目录
mkdir 你的文件夹名
#删除文件
rm 要删除的文件
#删除非空目录(谨慎使用!!,尤其是前面加sudo的!)
rm -rf /path/to/dir
#删除空目录
rmdir empty_dir
#运行bash脚本(bash脚本简单来说就是将比较长的linux命令写进.sh后缀的文档),如你现在在的目录下有install.sh脚本,运行需要输入:
bash install.sh
第二章:科学上网与Conda环境配置
2.1 为什么需要科学上网?
做生信分析,你会经常需要访问:
- GitHub
:下载各种工具和代码 - Bioconductor
:R语言的生信包仓库 - PyPI
:Python包仓库 - 各种数据库
:GEO、ArrayExpress等
这些网站有时候直连会很慢,甚至打不开。所以,配置一个稳定的网络环境很有必要。
2.2 网络工具的配置
提示:网络工具的具体配置方法还是需要自行了解,这里提供主要思路。
基本配置思路:
获取订阅链接(这一步需自行解决) 导入到工具(如clash)中 开启系统代理 选择合适的节点
验证是否成功:
打开浏览器,访问 google.com,能打开就OK!
喵喵师兄的进阶Tips (针对WSL用户):
如果你在WSL里下载东西慢,那是因为WSL默认不走Windows的代理。你需要配置WSL的网络代理,或者直接在.condarc里配置镜像源(见下文)。
推荐:使用clash-for-linux-install在WSL里快速安装Clash
nelvko/clash-for-linux-install: 😼 优雅地使用基于 clash/mihomo 的代理环境
在这里查看教程。订阅链接需要机/场,这部分需自己搞定
2.3 Anaconda:Python环境管理器
什么是Conda?
假如你目前有几个项目在跑:
项目A需要Python 3.8 + numpy 1.19 项目B需要Python 3.10 + numpy 1.24 项目C需要Python 3.9 + tensorflow 2.x
如果都装在一起,版本冲突问题极难处理。这时候就需要Anaconda来隔离这些不同的虚拟环境。
安装Miniconda(推荐)
注意,这里需要打开clash并开启全局代理
打开wsl(win+R,输入cmd,进入后输入wsl),输入
#创建并进入你希望下载的目录
mkdir /mnt/d/anaconda_wsl
cd /mnt/d/anaconda_wsl
#下载miniconda
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
#安装
bash Miniconda3-latest-Linux-x86_64.sh
按照提示操作:
按 Enter 阅读条款。 输入 yes 同意。 提示安装位置时,默认即可。 最后询问是否执行 conda init 时,务必输入 yes。 重启终端: 安装完成后,关闭并重新打开 WSL,你就会看到命令行前出现了 (base) 字样。 
验证安装:
conda --version
2.4 提速神器:Mamba 与 镜像源配置
很多同学抱怨 Conda 安装包太慢了,一直在 solving environment 转圈圈。
这里师兄教大家两个提速秘籍:
秘籍一:换国内镜像源 (如果不方便科学上网)
即使你有代理,有时候配置镜像源也会更快。
在命令行输入以下命令,把清华源加进去:
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
conda config --set show_channel_urls yes
注意:如果你开了全局代理,有时候反而需要把这些源去掉(用回官方源)。如果报错,尝试用 conda config --remove-key channels 恢复默认。
秘籍二:使用 Mamba (强烈推荐!)
Mamba 是 Conda 的C++重写版,速度快10倍以上!
# 在 base 环境下安装 mamba
conda install -n base -c conda-forge mamba
以后安装包,把 conda install 换成 mamba install,体验飞一般的感觉!
秘籍三:打开clash科学上网
如上面所写,这个方式我常用。因为国内镜像源我老是配出问题来)
2.5 创建单细胞分析环境
现在,让我们创建一个专门用于单细胞分析的环境!
第一步:创建新环境
# 使用 python 3.10 ,创建一个名为singlecell的conda环境
conda create -n singlecell python=3.10
第二步:激活环境
conda activate singlecell
激活后,你会看到命令行前面出现 (singlecell)。
第三步:安装单细胞分析必备包
这里我们一次性装好 Scanpy 和 Omicverse 所需的包。
注意: 如果你要用 Omicverse,建议先安装 PyTorch (见下篇)。这里我们先装基础包。
# 安装scanpy及其依赖
pip install scanpy
# 安装其他常用包
pip install leidenalg # 用于聚类
pip install harmonypy # 用于批次校正
验证安装:
python -c "import scanpy as sc; print(sc.__version__)"
第三章:IDE选择与配置
3.1 什么是IDE?
IDE(Integrated Development Environment,集成开发环境)就是写代码的工具。你可以理解为"Word是写文档的,IDE是写代码的"。
对于Python单细胞分析,主流的IDE选择有:
| Jupyter Lab/Notebook | ||
| VS Code | ||
| PyCharm |
喵喵师兄的推荐:
- 首选:VS Code + Jupyter插件
(有条件的可以把vscode替换成Cursor/Codex/TRAE/Qoder这样的,基于VS code的Vibe coding IDE,让AI自己帮你写代码改代码,国内的话Qoder比较方便) 备选:JupyterLab(纯Web界面) 现在还有Positron,跟Rstudio很像的新一代IDE,但是完全可以用来跑python
3.2 方案一:安装JupyterLab
JupyterLab是Jupyter Notebook的升级版,界面更现代,功能更丰富。
第一步:确保你在正确的conda环境中
conda activate singlecell
第二步:安装JupyterLab
pip install jupyterlab
第三步:在你的conda环境安装并注册Jupyter kernel(创建新环境时安装一次就行)
conda install ipykernel
python -m ipykernel install --user --name=singlecell --display-name singlecell
第三步:启动JupyterLab
jupyter lab
如果你用windows cmd:浏览器会自动打开你会看到一个类似文件管理器的界面。点击"Python 3"就可以新建一个Notebook开始写代码了!
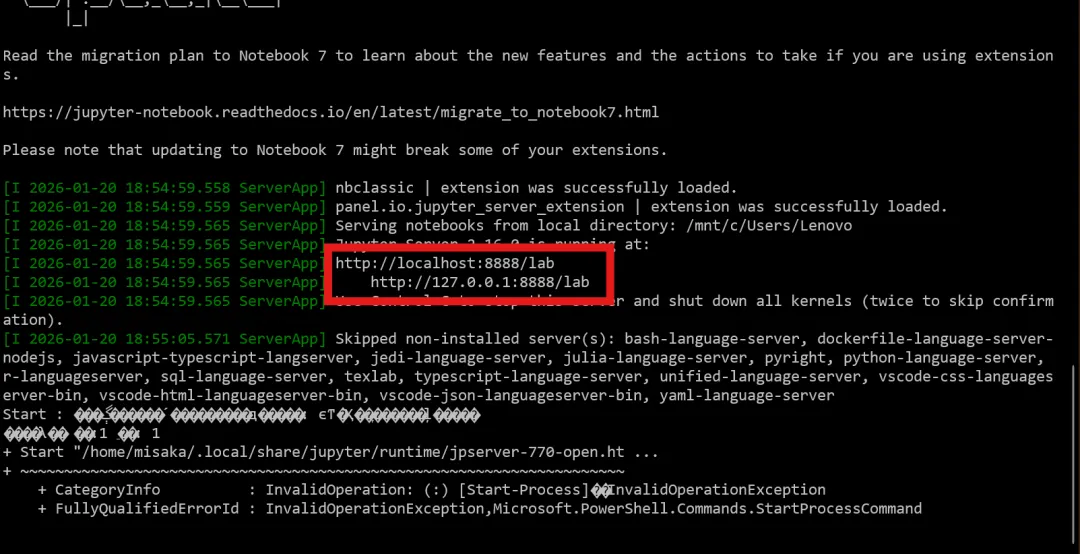
如果你用wsl/远程连接linux
会跳出来一堆东西,这时候在里面复制这个网址(http://localhost:8888/lab)到浏览器打开即可(你也可以找教程配置成自己打开,这里不赘述了)。打开后可能会让你设置或输入密码,设一个简单的就好。
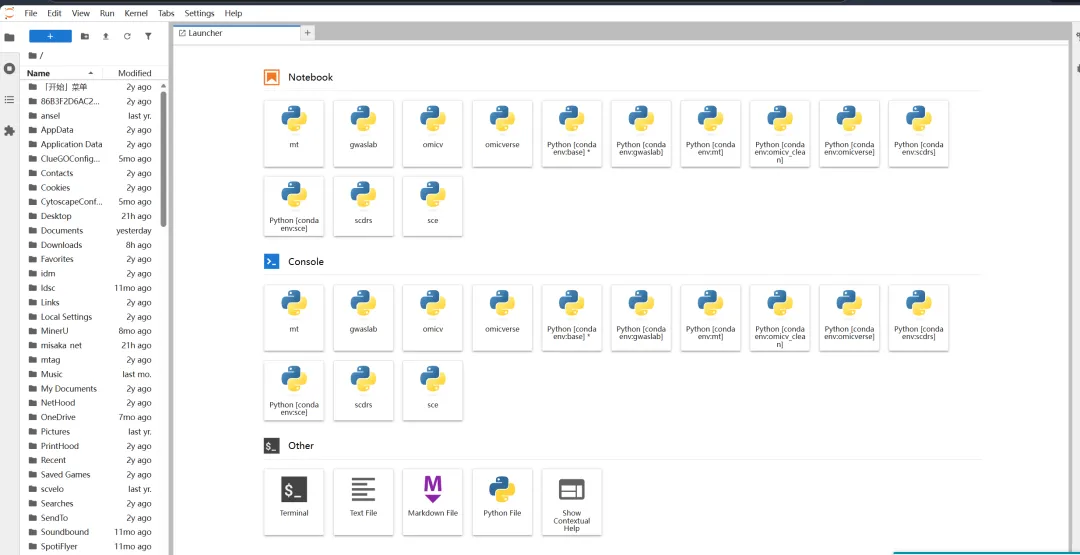
然后你就可以看到你创建的所有conda或者python环境啦:
注意:一般先在cmd/wsl里进入你需要的工作目录(cd /你的工作目录文件夹)再打开jupyter,比如要是你在C盘进入了Jupyter,是无法打开D盘的文件的。然后你在你的工作目录里无法访问这个目录的上级目录的
然后我们就可以创建一个新的.ipynb文件开始愉快进行python编程了!
记得看看右上角的环境是不是你要的 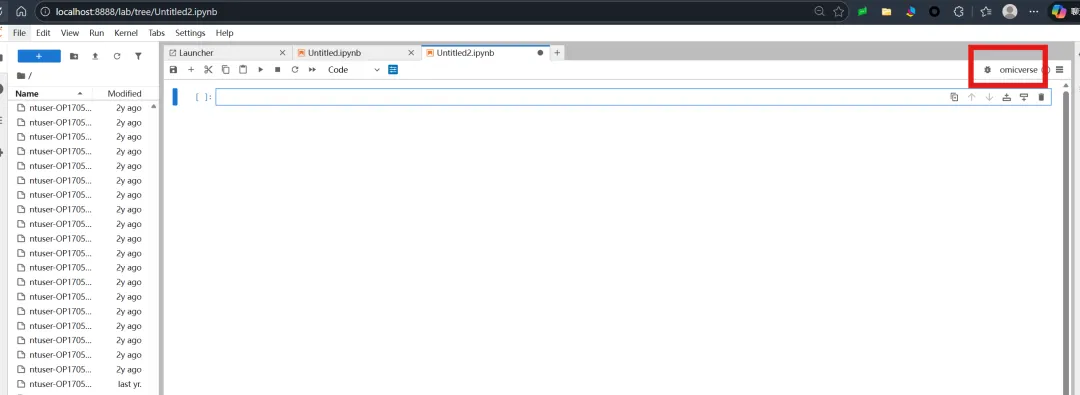
在里面的单元格输入代码,Ctrl + Enter即可运行,跟Rstudio很像
一些常用快捷键:
Shift + Enter:运行当前单元格并跳到下一个 Ctrl + Enter:运行当前单元格,光标不动 Esc + A:在上方插入新单元格 Esc + B:在下方插入新单元格 Esc + DD:删除当前单元格
3.3 方案二:VS Code(强烈推荐!)
VS Code是微软出品的免费代码编辑器,轻量又强大,堪称"宇宙第一编辑器"。
第一步:下载安装VS Code
如果你用的是windows CMD:官网:https://code.visualstudio.com/
下载后一路安装即可。
如果你使用wsl: 下载安装同第一步,在下一步安装好插件后,输入code .即可自动打开。
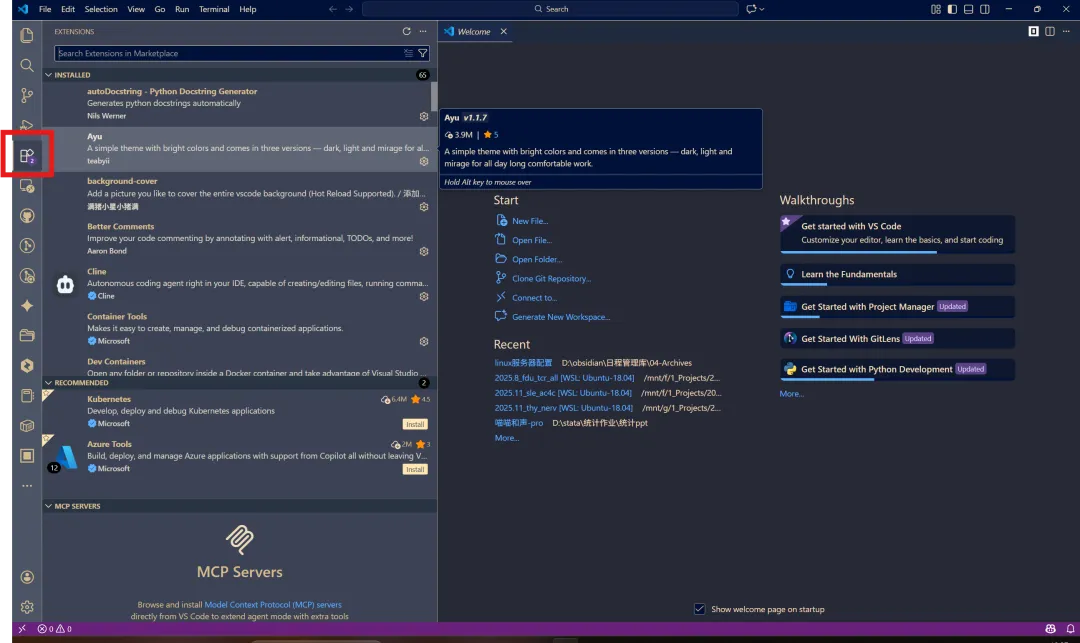
第二步:安装必备插件
VS Code 本身只是一个编辑器,它的强大在于“插件”。 打开 VS Code,点击左侧活动栏的 扩展图标(四个方块组成的图标,快捷键 Ctrl+Shift+X),在搜索框输入并安装以下插件:
- Python
:必装!提供代码高亮、智能提示和调试功能。 - Jupyter
:必装!让你能在 VS Code 里直接运行 .ipynb笔记本文件。 - WSL
:使用WSL的需要安装! 让你能流畅地在 Windows 上编辑和运行 Linux 子系统里的代码。 R也可以安装一下,如果你想以后在vscode写R代码
第三步:配置Python解释器
按 Ctrl + Shift + P,打开命令面板输入 Python: Select Interpreter选择我们之前创建的 scanpy环境
第四步:创建Jupyter Notebook
按 Ctrl + Shift + P输入 Jupyter: Create New Blank Notebook选择 scanpy环境作为内核开始愉快地写代码!
简单介绍下vscode的使用方法!
很多用习惯Rstudio的研究者第一次使用vscode都会有点懵,我这里来一步步教会大家如何使用VS code。
首先,在打开vscode时你会看见如下的界面:
要在VS code里跑代码,你首先需要打开你工作的文件夹,教程如下:
1. 界面布局对应
VS Code 的界面主要由侧边栏 (Side Bar) 和 编辑区 (Editor) 组成。
**资源管理器 *:点击左侧最上面的“文件夹”图标。
对应 RStudio 的右下角 Files 面板。这里管理你的项目文件。
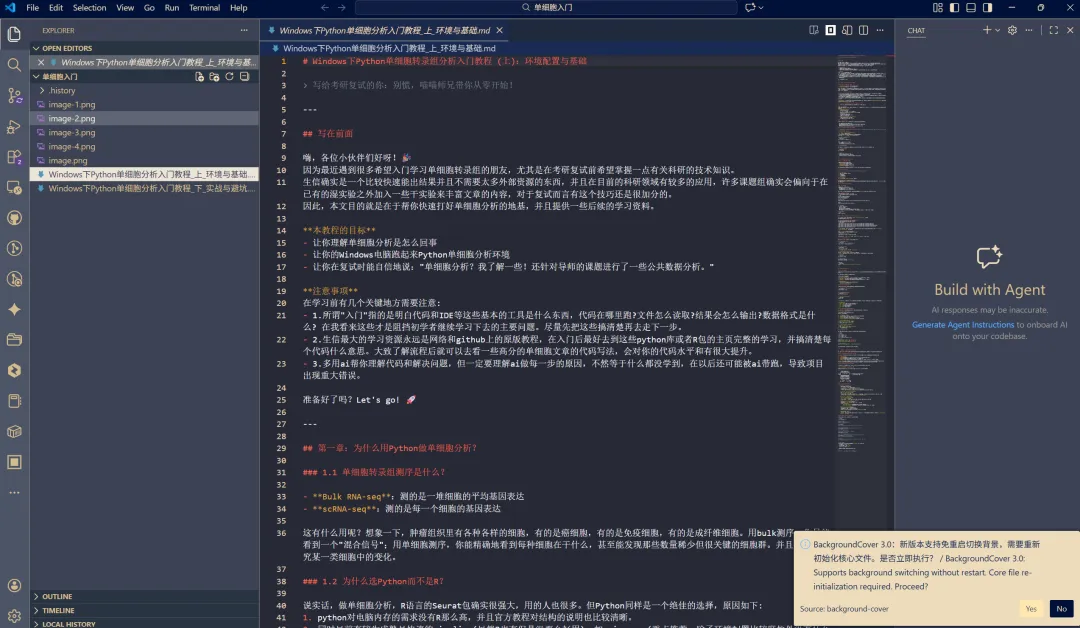
重要习惯:VS Code 是基于“文件夹”工作的。开始项目前,务必点击菜单栏
File -> Open Folder...打开你的项目文件夹,而不是只打开单个文件。来看一下比如我打开一个文件夹,并打开我的代码(我打开的是.md文件,但不管是你打开.py还是.R都是一样的!):
编辑器 (Editor Group):中间最大的区域。
- 对应 RStudio 的左上角 Source 面板
。这是你写代码的主战场。 大纲 (Outline):在资源管理器下方。
- 对应 RStudio 的右上角 Environment/History 下方的大纲
。可以快速跳转代码段落。 如何打开Jupyter?
右上角 files,新建并打开一个jupyter notebook,就跟打开jupyter一样啦!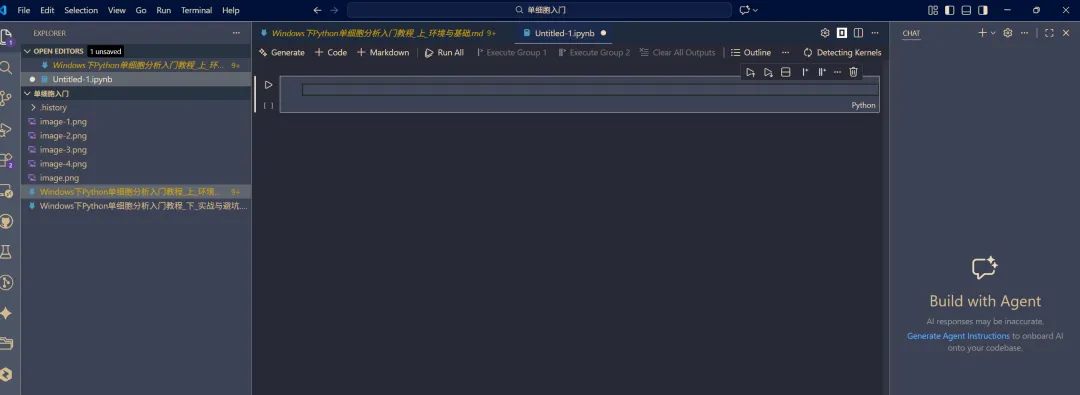
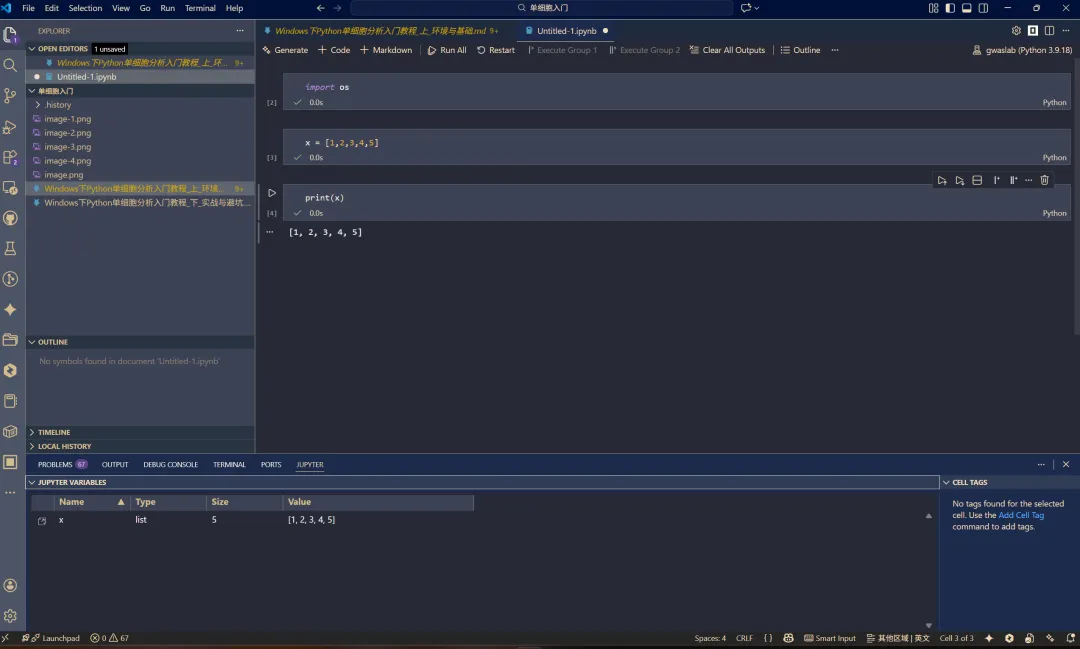
2. RStudio 用户最关心的:变量去哪了?(Environment)
在 RStudio 中,我们习惯随时查看右上角的 Environment 面板来确认数据框的维度和内容。在 VS Code 的 Jupyter 模式下,这个功能不仅有,而且更强大。
当你在 .ipynb 文件中运行了代码后,在terminal(如果没有我图里所示的terminal,左上角新建一个)里有一个jupyter,点击就能换看见环境变量了
3. 绘图与输出 (Plots & Console)
- 结果输出
:代码的运行结果(包括文本打印和静态图片)会直接显示在每个代码单元格(Cell)的下方。这比 RStudio 分离的 Console 更加直观,代码和结果紧密结合。 - 交互式绘图
:生成的图片支持缩放、保存为 PNG/SVG。点击图片左侧的“展开”图标,可以将图片在单独的窗口中查看。
4. 常用快捷键
| 运行当前格 | Shift + Enter | Shift + Enter | |
| 仅运行当前格 | Ctrl + Enter | Cmd + Enter | |
| 命令面板 | Ctrl + Shift + P | Cmd + Shift + P | |
| 注释代码 | Ctrl + / | Cmd + / | |
| 保存文件 | Ctrl + S | Cmd + S |
3.4 验证环境配置
无论你用JupyterLab还是VS Code,都在Notebook里运行以下代码来验证:
# 测试导入
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# 查看版本
print(f"Scanpy version: {sc.__version__}")
print(f"Pandas version: {pd.__version__}")
print(f"NumPy version: {np.__version__}")
# 测试scanpy功能
print("\n环境配置成功)
如果看到版本号输出,恭喜你,环境全部配置完成!
第四章:必须掌握的核心——Scanpy与AnnData
这一章是重点中的重点!如果不理解AnnData的结构,后面写代码就会像"瞎子摸象",经常报错都不知道错在哪。
4.1 Scanpy:Python单细胞核心库
Scanpy (Single-Cell Analysis in Python) 是目前Python生态中最主流、最权威的单细胞分析工具包。
为什么它这么强?
- 快
:处理100万个细胞的数据,R语言可能已经崩了,Scanpy还能跑得飞起。 - 全
:从读数据、质控、降维、聚类到画图,一条龙服务。 - 生态好
:现在最新的算法(比如去批次、轨迹推断、空间转录组)基本上都是基于Scanpy开发的。
复试加分项:如果老师问你"Scanpy和Seurat的区别",你可以回答:"Seurat是R语言的标准,适合统计分析;Scanpy基于Python,在处理超大规模数据(Large-scale data)和结合深度学习(Deep Learning)方面更有优势。"
4.2 AnnData:单细胞数据的"集装箱"
在Scanpy中,所有的数据都装在一个叫 AnnData (Annotated Data) 的对象里。
要理解Anndata,我们先来回忆一下普通bulk转录组的表达矩阵:
行为基因名,列为样本名。那么在anndata里,这个表达矩阵是什么样子呢?
首先我们来看这张Anndata的结构图,请务必把下面这张图刻在脑子里!Getting Started 你可以到这个网页去看看官方教程。
你可以把AnnData想象成一个"超级Excel表格",它由几个关联的部分组成:
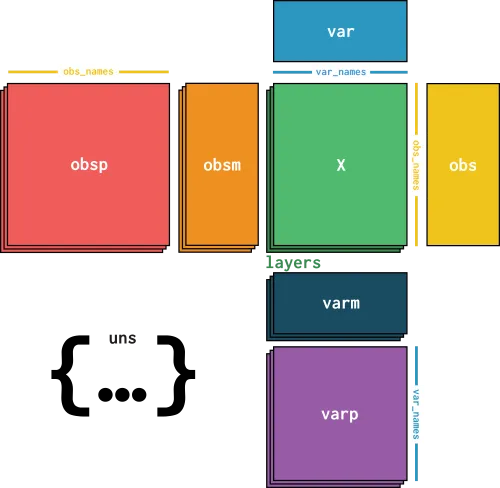
这张图怎么看呢?我们来理解一下:
我们把绿色的.X作为核心,它就是类似上面转录组的那个表达矩阵,但因为是矩阵,其中仅存储着数字,也就是中间的表达谱。
而上面的列名var_names就是基因名,行名obs_names则是细胞名。这是整个adata的最核心部分,详细说明如下:
拆解AnnData的五脏六腑:
adata.X(核心矩阵)这就是你的测序数据本体。 - 行 (Rows)
= 细胞 (Observations, n_obs) - 列 (Columns)
= 基因 (Variables, n_vars) 里面的数值通常是 counts(计数) 或者经过标准化的数值。- 注意
:为了省内存,这里通常存的是"稀疏矩阵"(Sparse Matrix),只存非0的值。 adata.obs(细胞信息表 —— 重点!)这是一个 pandas.DataFrame。- 每一行对应一个细胞
。 用来存细胞的"身份证信息":比如这个细胞属于哪个样本?也是哪种细胞类型(Cluster)?线粒体含量是多少? - 以后你做的聚类结果(比如 'leiden' 列),都会存在这里!
adata.var(基因信息表)也是一个 pandas.DataFrame。- 每一行对应一个基因
。 用来存基因的属性:比如基因名(Symbol)?是不是高变基因(highly_variable)? adata.obsm(细胞的多维数据 —— 难点!)adata.obsm['X_pca']:存PCA降维后的坐标(比如前50维)。 adata.obsm['X_umap']:存UMAP的坐标(2维),画图的时候其实就是画这几个数! 这里存的是矩阵,不是单列数据。 关键特征:行数 = 细胞数 (必须和细胞一一对应)。 - 最常见的例子
: adata.uns(杂七杂八的信息)Unstructured data(非结构化数据)。 只要不跟细胞/基因一一对应的,都扔这儿。 比如:计算邻接图的参数、聚类用的颜色盘( leiden_colors)等。adata.layers(图层)就像PS的图层一样。 adata.X只有一份,但如果你想同时保存原始counts和标准化后的数据/scale后的数据,就可以把scale后的数据塞进 adata.layers['scale'] 里备份。
4.3 实战:像做手术一样解剖数据
光说不练假把式,我们在代码里看看它到底长啥样。
import scanpy as sc
# 读取示例数据
adata = sc.datasets.pbmc3k()
# 1. 看看整体长相
print(adata)
AnnData objectwith n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
Empty DataFrame
Columns: []
Index: [AAACATACAACCAC-1, AAACATTGAGCTAC-1, AAACATTGATCAGC-1, AAACCGTGCTTCCG-1, AAACCGTGTATGCG-1]
gene_ids
index
MIR1302-10 ENSG00000243485
FAM138A ENSG00000237613
OR4F5 ENSG00000186092
RP11-34P13.7 ENSG00000238009
RP11-34P13.8 ENSG00000239945
[[0.0.0.]
[0.0.0.]
[0.0.0.]]
# 2. 看看细胞信息 (obs)
# 刚读入时可能只有索引,后面做完分析这里会多出很多列
print(adata.obs.head())
Empty DataFrame
Columns: []
Index: [AAACATACAACCAC-1, AAACATTGAGCTAC-1, AAACATTGATCAGC-1, AAACCGTGCTTCCG-1, AAACCGTGTATGCG-1]
# 3. 看看基因信息 (var)
# 这里通常存着 Gene Symbol 和 Gene ID
print(adata.var.head())
gene_ids
index
MIR1302-10 ENSG00000243485
FAM138A ENSG00000237613
OR4F5 ENSG00000186092
RP11-34P13.7 ENSG00000238009
RP11-34P13.8 ENSG00000239945
# 4. 重点:查看数据矩阵 (X)
# 因为是稀疏矩阵,直接print看不了,要用toarray()转成普通数组
print(adata.X[:3, :3].toarray())
# 输出前3个细胞、前3个基因的表达量
[[0.0.0.]
[0.0.0.]
[0.0.0.]]
# 喵喵Tip:
# 在分析过程中,随时 print(adata) 看看你的 obs 和 var 多了什么列
# 是排查bug的最好方法!
总结一下:
做单细胞分析,本质上就是不断地往这个 AnnData 对象里填东西:
算完QC指标 -> 填进 obs找完高变基因 -> 填进 var算完PCA/UMAP -> 填进 obsm聚完类 -> 结果填回 obs
搞懂了这个,你就搞懂了Scanpy的半壁江山!
这篇也讲的差不多了,对于一些python和Scanpy/Omicverse的使用,会在下一篇进行更新。下一期会着重讲解Scanpy使用以及很好用的Omicverse库的使用(虽然还是更推荐大家直接去看官方教程)
希望大家都能熟练掌握使用python/R进行单细胞分析!尤其在现在有AI的帮助下,学习这些工具并不是一件难事,现在就开始试试吧 ~
觉得写的好可以关注一下博主哦~~

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 一顿饭能讲出多少故事?用 Python 分析“小费数据”,发现人性真相!
- 网安运维必知会:7类Linux命令解决80%问题
- 场景实操 D1(21 天 D6)|代码报错求助:日企 IT 对前辈的日语话术模板
- 166 个最常用的 Linux 命令汇总,总有你需要用到的!
- Python反序列化:谁说 Python 只有 pickle RCE?
- 今年读的这本书我愿称之为Python量化大杀器
- 看完这1456页,我跪了!Linux系统管理员之路,有这一份笔记就够了!
- Linux磁盘空间满了怎么办?这套排查流程救了我无数次
- 终于把python做成了编程软件
- MX Linux 再次“逆流而行”:25.1 版本重新拥抱双 init
