浙江大学医学院附属第一医院王雪玉教授和德国杜伊斯堡-埃森大学医学院病毒研究所陆蒙吉教授团队在《Virologica Sinica》期刊发表了题为“HBV and host metabolic crosstalk: Reprogramming pathways for viral replication and pathogenesis”的综述文章,系统阐述了乙型肝炎病毒(HBV)如何操纵宿主代谢以维持其生命周期并促进疾病进展的机制。
摘要:乙型肝炎病毒 (HBV) 通过劫持宿主代谢网络建立慢性感染,驱动一系列肝脏病理,从肝炎到肝硬化和肝细胞癌。从机制上讲,HBV 重编程核心代谢通路,包括糖酵解、三羧酸 (TCA) 循环、氧化磷酸化和脂质稳态,为其复制机器提供燃料并逃避免疫监视。本综述系统性地总结了当前关于 HBV 诱导的葡萄糖/脂质代谢重编程的证据,特别强调在代谢层面的病毒-宿主相互作用如何调控病毒的发病机制。
关键词:乙型肝炎病毒 (HBV);代谢重编程;糖酵解;三羧酸循环;氧化磷酸化;脂质代谢
引言
肝脏作为身体的主要代谢枢纽,通过对碳水化合物、脂质和氨基酸代谢的精确调控来协调全身稳态。这种代谢优势,由肝细胞(占肝脏质量的80%)中复杂的转录网络所支配,源于其独特的内质网 (ER) 和线粒体丰度 (Rui, 2014; Gissen and Arias, 2015)。虽然这些特性对生理平衡至关重要,但它们也使肝脏容易受到病理上的利用——特别是在慢性病毒性肝炎感染期间,代谢重编程驱动疾病进展。
HBV 是嗜肝DNA病毒科的一种小型包膜DNA病毒,主要感染肝细胞并在全球数百万人中建立慢性感染,是肝硬化和肝细胞癌 (HCC) 的主要原因 (Hsu et al., 2023)。在感染者的血清中,它以三种颗粒形式存在:含有rcDNA聚合酶复合物的42纳米感染性Dane颗粒,以及非感染性的22纳米球形和丝状颗粒。病毒包膜包含三种共末端的糖蛋白——小(S)、中(M)和大(L)表面抗原——它们介导病毒的进入和组装。HBV 主要利用 ER-高尔基体-内体途径进行颗粒分泌,而持续感染诱导的 ER 应激促进了自噬体介导的病毒成分的替代运输 (Wang, X. et al., 2022a; Wang, X. et al., 2022b)。虽然抗病毒治疗可以抑制 HBV 复制,但由于受感染细胞核中共价闭合环状 DNA (cccDNA) 的持续存在,它们无法根除病毒 (Hu and Huang, 2024)。
HBV 策略性地劫持肝脏代谢以促进复制并逃避免疫 (Bard-Chapeau et al., 2014; Xie et al., 2017; Diaz et al., 2022; Willmann and Moita, 2024)。临床代谢组学研究揭示,在疾病进展过程中,三羧酸 (TCA) 循环和脂质通路逐渐失调 (Yang et al., 2016; Li J. et al., 2022),而 HBV 表面抗原 (HBsAg) 血清清除则与抑制的糖酵解/糖异生基因活性、增强的脂肪酸 (FA) 降解以及升高的 TCA 代谢物相关 (Lin et al., 2025)。从机制上讲,HBV 通过病毒蛋白与宿主酶的直接相互作用破坏肝脏葡萄糖稳态。HBV X 蛋白 (HBx) 和 HBsAg 在转录和翻译后水平调控糖酵解和糖异生中的关键限速酶,如 PKM2 和磷酸烯醇式丙酮酸羧激酶 (PEPCK) (Wang and Zhang, 2023),迫使肝细胞转向“瓦博格效应”来富集病毒复制所需的 ATP 和原材料。这种调控通过肝脏富集的转录因子得到进一步加强,这些转录因子通过与病毒启动子中存在的特定结合位点直接调节 HBV 活性 (Turton et al., 2020)。此外,通过病毒基因组整合产生的截短型 HBx 蛋白进一步扰乱了氧化还原平衡,创造了一个有利于病毒持续存在的微环境 (Zhang, Y. et al., 2021)。脂质代谢同样以阶段特异性的方式被劫持:早期感染促进脂滴形成用于衣壳组装,而肝硬化阶段则抑制胆固醇合成 (Arain et al., 2018; Zhang, J. et al., 2021)。值得注意的是,仅 HBx 本身就足以改变肝细胞的脂质谱 (Lamontagne et al., 2018)。此外,营养物质的可用性通过自噬介导的过程动态调节 HBV 的产生,揭示了宿主代谢通路与病毒传播之间复杂的相互作用。
值得注意的是,这些代谢重塑事件表现出时空异质性,并与病毒生命周期的不同阶段精确耦合。然而,目前仍缺乏对控制 HBV 诱导的代谢重编程及其与宿主免疫微环境相互作用的动态调控网络的全面、系统性分析。本综述旨在整合临床数据与分子机制研究,总结 HBV 代谢相互作用的多层次调控框架,并讨论关键代谢分子作为创新治疗靶点的潜力。
HBV 劫持的代谢途径
葡萄糖代谢重编程
糖酵解通量放大(类瓦博格重编程)
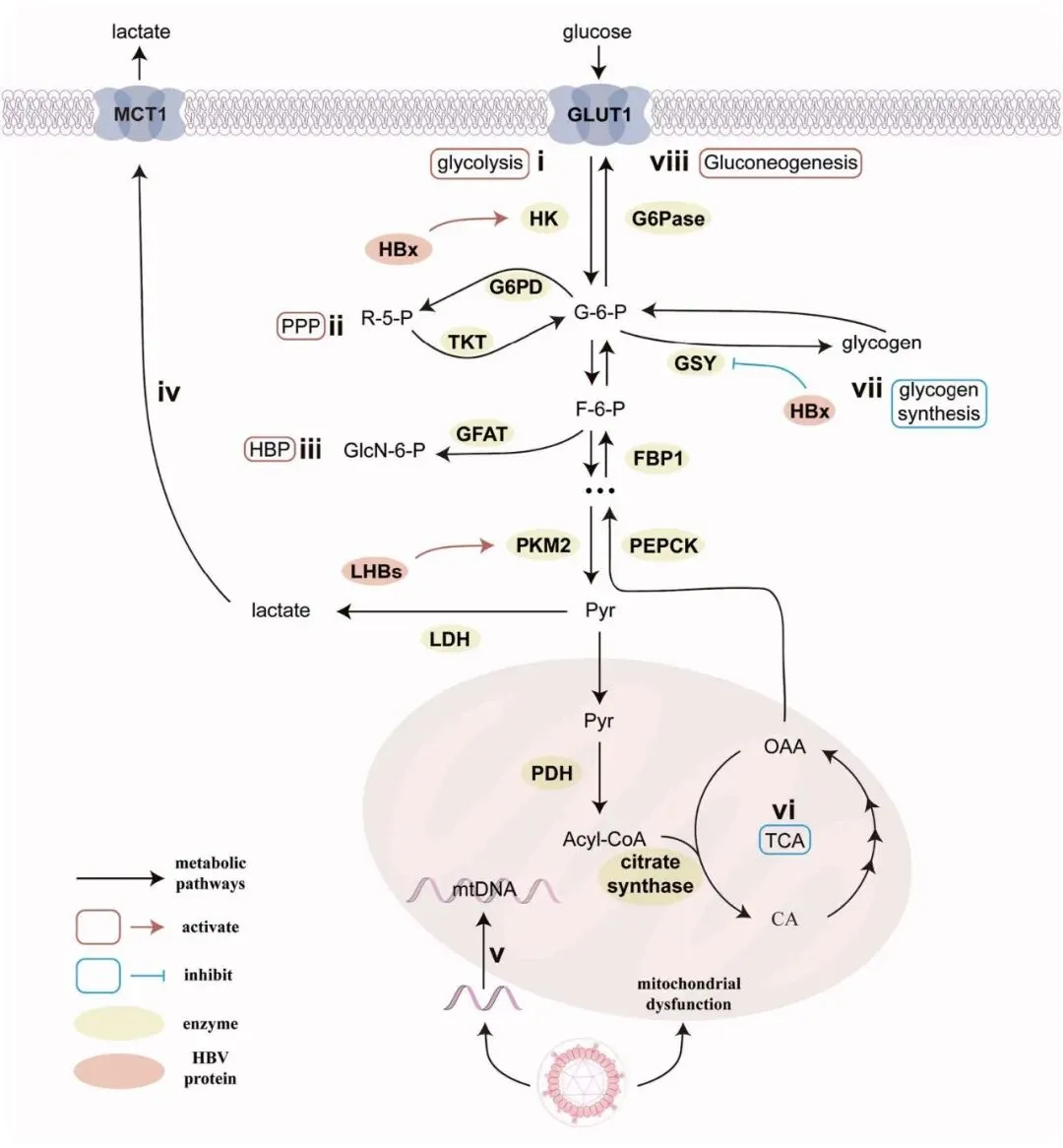
HBV 策略性地重编程宿主葡萄糖代谢以支持病毒复制和持续存在,主要通过劫持糖酵解(包括 TCA 循环)、磷酸戊糖途径 (PPP) 和己糖胺生物合成途径 (HBP)(图 1 i-iii)。在正常生理条件下,在有氧条件下,大部分葡萄糖经历氧化磷酸化 (OXPHOS) 在有氧条件下 (Cooper and Adams, 2009),但 HBV 感染诱导了类瓦博格转变——尽管有氧气可用,病毒诱导宿主细胞优先进行有氧糖酵解 (Koppenol et al., 2011; Dayton et al., 2016)。
乳酸积累主要损害肝脏免疫细胞的功能,从而建立有利于病毒持续存在的免疫抑制微环境。升高的乳酸水平不仅通过调节单核巨噬细胞、中性粒细胞、树突状细胞和自然杀伤 (NK) 细胞来损害先天免疫反应,还导致 CD8⁺ T 细胞功能障碍 (Colegio et al., 2014; Deng et al., 2021; Liu, H. et al., 2024; Plebanek et al., 2024; Jin et al., 2025)。值得注意的是,通过单羧酸转运蛋白 1 (MCT1) 输出的乳酸激活了磷脂酰肌醇 3-激酶 (PI3K)/蛋白激酶 B (AKT/PKB) 信号通路(图 1 iv)(Chen, L. et al., 2022),驱动 HBV 感染的肝细胞恶性增殖。此外,乳酸驱动的组蛋白乳酸化表观遗传地重编程代谢酶的基因表达,以维持有利于病毒持续存在的环境 (Zhang, D. et al., 2019; Gao et al., 2023; Jin et al., 2023; Yang, Z. et al., 2023)。
HBV 通过 miRNA 介导的调控进一步微调这种代谢格局。HBV 利用促病毒 miRNAs(例如 miR-30b-5p)来增强支持感染肝细胞增殖的糖酵解通量 (Chen, W. et al., 2021)。而宿主对策则利用靶向关键代谢调节因子的抗病毒 miRNAs(例如 miR-130a),如过氧化物酶体增殖物激活受体 γ 共激活因子 1-α (PGC1α)、过氧化物酶体增殖物激活受体 γ (PPARγ) 以及在肝脏和红细胞中编码丙酮酸激酶的基因 (PKLR) (Huang, J. Y. et al., 2015; Duan et al., 2018),以恢复代谢平衡并限制病毒复制。这种复杂的相互作用凸显了 HBV 在逃避免疫监视的同时利用宿主代谢的能力。
己糖胺生物合成途径和磷酸戊糖途径:葡萄糖代谢旁路激活
HBV 感染策略性地激活宿主葡萄糖代谢旁路以支持病毒复制和致癌过程。这种代谢重编程主要涉及两个关键途径:HBP,它参与蛋白质糖基化并调节细胞信号以调控 HBV 复制;以及 PPP,一个关键的葡萄糖代谢旁路,为核苷酸合成提供核糖,并产生烟酰胺腺嘌呤二核苷酸磷酸 (NADPH) 以支持还原性生物合成和抵抗氧化应激。
1). HBP。一项关键研究表明,HBV 上调葡萄糖转运蛋白 1 (GLUT1) 和谷氨酰胺-果糖-6-磷酸酰胺转移酶 (GFAT) (Li, H. et al., 2015),这是 HBP 的两个关键酶,增加了 UDP-GlcNAc——O-GlcNAc 修饰的底物——的产量。这种翻译后修饰产生双重效应:RNA N⁶-甲基腺苷 (m6A) 阅读蛋白 YTH 结构域 2 (YTHDF2) 的 O-GlcNAc 修饰促进 HBV 复制和肝癌进展 (Yang, Y. et al., 2023);而包含无菌α基序和组氨酸/天冬氨酸结构域的蛋白 1 (SAMHD1,一种病毒限制因子) 的 O-GlcNAc 修饰抑制其磷酸化,反而抑制 HBV 活性 (Hu et al., 2021)。值得注意的是,药理学抑制 HBP 对 HBV 感染和肝癌发生产生矛盾的影响。我们之前的研究揭示,用小分子抑制剂 (OSMI-1) 阻断 HBP 尽管降低了 UDP-GlcNAc 水平,却意外地增强了 HBV 复制和基因表达 (Wang, X. et al., 2020)。从机制上讲,HBP 抑制引发了 ER 应激,激活了未折叠蛋白反应 (UPR) 通路,进而上调了病毒转录。此外,HBP 抑制通过阻断自噬体-溶酶体融合损害了自噬流,导致缺陷自噬体积累,这些自噬体可能为 HBV 组装提供膜支架并与膜融合以利于病毒释放。有趣的是,虽然 HBP 抑制促进了病毒的持续存在,但它同时通过减少致癌信号分子(例如 eEF1A1, YTHDF2)的 O-GlcNAc 糖基化修饰来抑制肝细胞癌进展 (Yang, Y. et al., 2023; Zhou et al., 2023)。这表明 O-GlcNAc 修饰在平衡病毒适应性和致癌作用中具有环境依赖性作用。
2). PPP,另一个关键的葡萄糖代谢旁路,通过葡萄糖-6-磷酸脱氢酶 (G6PD) 和转酮醇酶 (TKT) 产生 NADPH 和核糖-5-磷酸,满足 HBV 的生物合成需求 (Liu, Qi et al., 2022)。在 HBV 相关的 HCC 中,G6PD 和 TKT 均升高,反映了核苷酸合成增强 (Kittaka et al., 2010)。从机制上讲,HBx 激活 NF-E2 相关因子 2 (Nrf2),从而促进 G6PD 表达、抗凋亡因子 (Bcl-2, Bcl-xl) 和细胞周期蛋白的表达 (Liu, B. et al., 2015)。此外,HBx 刺激信号转导与转录激活因子 3 (STAT3) 在 Tyr-705 位点的磷酸化,并通过 SH2D5-TKT 相互作用促进 STAT3 的活性及下游信号通路,加速核苷酸合成,并促进肝细胞癌增殖 (Zheng, Y. et al., 2019)。
总之,HBV 构建了一个复杂的代谢重编程网络,利用 HBP 和 PPP 来优化病毒复制,同时创造一个促肿瘤微环境。O-GlcNAc 修饰和 G6PD-Nrf2-STAT3 轴的双重作用突显了病毒持续存在与宿主代谢适应性之间的微妙平衡,为破坏 HBV 相关致癌作用提供了新的治疗靶点。
线粒体重编程策略
在正常生理条件下,TCA 循环作为线粒体代谢的中心枢纽,为 ATP 生产提供燃料并产生还原当量以维持细胞能量需求 (Arnold and Finley, 2023)。然而,HBV 颠覆了这些典型的代谢途径,以满足其复制和分泌的高度需求,驱动了线粒体葡萄糖代谢的广泛重编程(图 1 vi)(Vander Heiden et al., 2009; Li, Y. and Ou, 2023)。在 HCC 中,细胞甲基化测序揭示,HBV 诱导的表观遗传修饰主要破坏了三个相互关联的代谢过程:糖酵解、TCA 循环和 OXPHOS (Ye et al., 2016)。值得注意的是,HBV 优先将其遗传物质整合到对能量生产至关重要的位点的线粒体 DNA (mtDNA) 中,例如细胞色素 C 氧化酶 III,一个关键的 OXPHOS组件(图 1 v)(Oikawa et al., 2022)。这种整合破坏了 OXPHOS 效率,但矛盾地增强了糖酵解通量,从而将葡萄糖来源的碳重新导向以支持病毒复制 (Giosa et al., 2023)。这种代谢重编程不仅维持了 HBV 的传播,还通过调节宿主免疫细胞的代谢状态来削弱免疫监视。
HBV 与免疫细胞之间的代谢相互作用表现出极化依赖性动态。在促炎性 M1 型巨噬细胞中,HBV 上调 OXPHOS 以抑制 IL-1β 的表达,有效中和其抗病毒活性并促进病毒持续存在 (Li, Y. et al., 2022)。相反,在抗炎性 M2 型巨噬细胞中,HBV 诱导线粒体酶的过度乙酰化,包括柠檬酸合酶和丙酮酸脱氢酶复合物 (PDH),以抑制丙酮酸衍生的乙酰辅酶 A 的产生,随后限制丙酮酸流入 TCA 循环 (Bei et al., 2023; Selvamani et al., 2024)。这种代谢限制可能触发代偿性谷氨酰胺回补以补充 TCA 中间体,这已被证明能进一步促进 HBV 转录和复制 (Raney et al., 1991; Murad et al., 2021; Cheng, S.T. et al., 2025)。HBV 进一步将其代谢操纵扩展到适应性免疫 (Wang, L. et al., 2023)。在活化的 CD8⁺ T 细胞中,HBV 感染导致线粒体功能障碍,使代谢从 OXPHOS 转向有氧糖酵解,从而损害效应功能和抗病毒反应 (Schurich et al., 2016)。类似地,HBV 相关肝硬化中的 B 细胞表现出线粒体功能受损,导致增殖和分化缺陷——这种代谢缺陷加剧了感染相关的并发症 (Huang, C. et al., 2021)。
HBV 精心策划了一个多方面的代谢劫持策略,靶向线粒体代谢以为其复制提供燃料,同时削弱宿主免疫防御。这种双重机制突显了调节代谢通路作为治疗途径的潜力,以破坏病毒持续存在并恢复慢性 HBV 感染中的免疫能力。
糖原代谢失调
糖原合成与利用
糖原主要储存在肝脏和肌肉中,是维持血糖稳态的关键储备。肝内糖原在维持血糖水平,特别是在空腹状态下,起着至关重要的作用。然而,在 HBV 感染及其进展为 HCC 的背景下,发生了代谢转变,其特征是从糖原储存转向糖原利用 (Toshkov et al., 1994)。这种转变伴随着糖酵解的增强。负责糖原合成的关键酶——糖原合酶 (GYS),在 HBV 相关的 HCC 中下调 (Chen, S.L. et al., 2019)。此外,HBV pre-S2 区域的突变可能触发糖酵解,从而促进糖原利用(图 1 vii)(Teng et al., 2015)。HBx 可能对糖原合成表现出抑制作用,从而破坏肝细胞终末分化并促进细胞增殖 (Huang, J. et al., 2012)。此外,HBx 通过激活 AKT/糖原合酶激酶 (GSK-3β)/β-连环蛋白信号通路促进肝癌发生 (Khattar et al., 2012),进一步促进肿瘤发生。糖原合酶激酶 3 (GSK3) 抑制剂可以抑制 HBV e 抗原 (HBeAg) 和 HBsAg 的产生 (Nishitsuji et al., 2025)。
糖异生途径
在空腹条件下,胰高血糖素与胰高血糖素受体 (GCGR) 结合上调关键的糖异生酶,包括 PEPCK 和葡萄糖-6-磷酸酶 (G6Pase)。值得注意的是,小乙型肝炎病毒表面抗原 (SHBs) 已被证明通过直接结合腺苷酸环化酶 (AC) 1 启动子来激活这些糖异生步骤 (Chen, Y. et al., 2022)。CREB 调节的转录共激活因子 2 (CRTC2) 在这个过程中扮演重要角色。当被激活时,CRTC2 通过诱导 PGC1α(糖异生的中心调节因子)来增强 HBV 转录和复制,从而在肝脏代谢信号与病毒生物合成之间建立直接联系 (Tian et al., 2014)。相反,磷酸烯醇式丙酮酸羧激酶 1 (PCK1),即 PEPCK 的线粒体亚型,通过抑制 PGC1α 的活性来抑制 HBV 复制(图 1 viii)(Tang et al., 2019)。HBx 通过与关键转录因子(包括 PGC1α)相互作用并调节核受体 (NRs)(如肝细胞核因子 4-α (HNF4α) 和叉头框蛋白 O1 (FOXO1))的表达,进一步调控肝脏代谢通路(图 2 viii)(Haviv et al., 1995; Reese et al., 2011; Tian et al., 2013)。这些转录因子不仅参与 HBV 启动子调控,也对病毒转录有显著贡献。短期禁食通过激活 PGC1α 增强肝脏糖异生,这反过来又诱导 HBV 表达 (Shlomai et al., 2006; Shlomai and Shaul, 2009)。尽管在水动力注射模型中,热量限制对 HBV 生命周期没有显著影响,但长期观察表明其效果可能随时间变得更加明显 (Li, L. et al., 2009)。因此,靶向 PGC1α 已被提议作为 HBV 治疗的潜在策略 (Jhuang et al., 2015),为调节病毒复制和宿主代谢通路提供了一个有前景的方法。
除了这些机制外,HBx 激活 iNOS 启动子的转录起始位点,启动一氧化氮 (NO)/c-Jun N-末端激酶 (JNK) 信号通路来激活糖异生,从而促进关键糖异生酶的表达,包括 PEPCK 和 G6Pase (Shin et al., 2011)。此外,果糖-1,6-二磷酸酶 1 (FBP1),另一个关键的糖异生酶,在 HBV 感染的背景下受到表观遗传调节(图 1 viii)(Sengupta et al., 2022)。总的来说,这些发现强调了 HBV 感染与肝脏糖异生代谢之间复杂的相互作用,突出了用于治疗干预的新代谢靶点。
脂质代谢重编程
HBV 感染中的脂肪酸代谢操纵
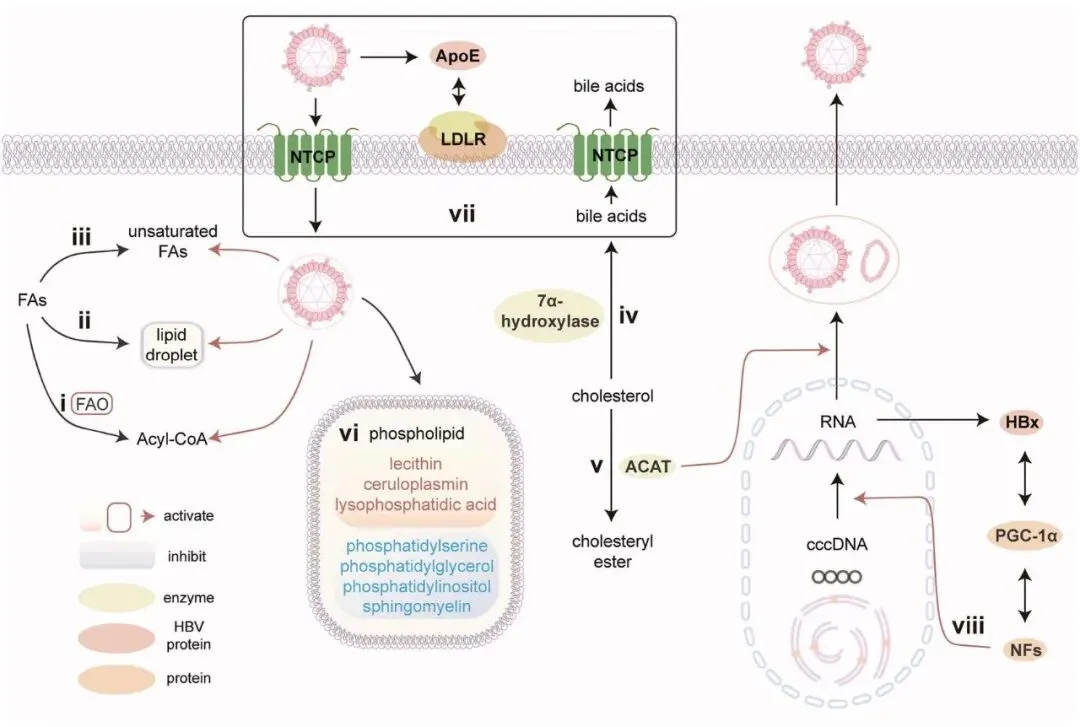
HBV 广泛地重编程宿主 FA 代谢,以建立一个有利于病毒复制和持续存在的环境。该病毒主要通过其调节蛋白 HBx 来调节 FA 代谢,HBx 协调代谢向脂肪酸氧化 (FAO) 和脂肪生成转变。HBx 促进钙动员到细胞质中,从而刺激 AMP 活化蛋白激酶 (AMPK) 以增强 FAO(图 2 i),使能量在营养压力下得以产生 (Wang, M.D. et al., 2016),同时上调脂肪生成因子甾醇调节元件结合蛋白 1 (SREBP1) 和 PPARγ 以驱动脂质积累和不饱和 FA 衍生物的合成(图 2 ii, iii)(Kim, K.H. et al., 2007; Shan et al., 2010)。该病毒与 FA 表现出复杂的、物种依赖性的关系:长链 FA(例如棕榈酸)稳定 HBx 并促进复制 (Okamura et al., 2016; Wu, Y.L. et al., 2016; Huang, J. et al., 2017; Zhang, W. et al., 2025),而短链 FA 表现出对 HCC 进展的保护作用 (Mcbrearty et al., 2021),外源性油酸则矛盾地抑制 HBV 分泌 (Liu, Qichuang et al., 2022)。HBV 进一步优化其 FA 环境,通过上调 FABP1 来保留促病毒的长链 FA (Wu, Y.L. et al., 2016),同时降低抗病毒的短链 FA 水平 (Zhang, W. et al., 2025)。此外,临床观察发现,增强的 FAO 是 HBV 相关 ACLF 的一个关键代谢特征,并表明用曲美他嗪抑制 FAO 可提高患者生存率 (Yu et al., 2020)。
从流行病学上看,慢性 HBV 感染与较低的肝脏脂肪变性患病率和更有利的血脂谱相关,这引发了对其潜在机制的疑问。这种“保护性”表型并非反映病毒的良性作用,而是源于涉及代谢重编程和免疫调节的病毒-宿主相互作用。Cheng 等人 (2023) 表明,HBV 单一感染者血清甘油三酯、胆固醇、LDL 和 HDL 降低,提示
HBV 抑制脂肪生成 (Cheng, Y.M. et al., 2023)。从机制上讲,HBx 可能破坏核受体信号(例如,PPARα, LXR),将脂质前体转向病毒复制。同时,HBV 调节免疫力:Huang 等人 (2024) 报告称,患有代谢功能障碍相关脂肪性肝病 (MASLD) 的慢性乙型肝炎 (CHB) 患者获得了更高的 HBsAg 清除率 (Huang, S.C. et al., 2024)。HBx 抑制炎症小体激活(例如 NLRP3),并且 HBV 驱动的乳酸积累损害 NK、树突状和 CD8⁺ T 细胞功能,助长免疫抑制。然而,这种明显的保护作用是矛盾的且具有环境依赖性。CHB 患者,尤其是那些 HBV DNA 水平高的患者,显示出脂肪变性减少 (Huang, S.C. and Liu, 2023; Liu, C.J. et al., 2024),但一旦发生 MASLD,代谢和病毒损伤协同作用加速纤维化。因此,HBV 最初可能通过代谢和免疫途径抑制脂肪变性,但代谢功能障碍最终会揭示并放大其致病潜力。HBV 与 FA 代谢之间这种复杂的相互作用不仅支持病毒复制,也有助于 HBV 相关肝病的进展,为治疗干预提供了新途径。
HBV 对胆固醇代谢的重编程
HBV 在其生命周期的多个阶段,特别是病毒进入和组装,严重依赖胆固醇,这一点在膜胆固醇耗竭后感染性显著降低中得到证明 (Bremer et al., 2009; Pollock et al., 2010)。最近的研究表明,抑制酰基辅酶 A:胆固醇酰基转移酶 (ACAT),一种胆固醇酯化的关键酶,具有三重治疗益处:直接抑制 HBV 颗粒生成,通过脂质代谢重编程(减少中性脂滴同时增强 TCR 信号传导)恢复耗竭的 T 细胞功能,并与程序性死亡受体 1 (PD-1) 阻断剂协同作用,重振抗病毒免疫力 (Schmidt et al., 2021)(图 2 v)。HBV 可能通过氧化的胆固醇衍生物来操纵胆固醇代谢,这些衍生物激活肝 X 受体 (LXR) 核受体以促进病毒转录 (Kim et al., 2011; Cui, M. et al., 2014)。矛盾的是,尽管 HBV 利用胆固醇进行复制,但恢复胆固醇稳态可以通过改善树突状细胞功能来增强抗病毒免疫力 (Zhao et al., 2022),突显了病毒需求与宿主防御之间复杂的相互作用。此外,HBV 通过上调胆固醇 7α-羟化酶(图 2 iv)和调节钠-牛磺胆酸共转运多肽 (NTCP) 受体(图 2 vii)(Geier, 2014; Oehler et al., 2014),深刻地改变了胆汁酸代谢,创造了额外的代谢弱点,这些弱点可能成为治疗靶点。
磷脂在 HBV 组装和入胞中的作用
越来越多的证据表明,HBV 广泛地重编程宿主磷脂代谢以促进其生命周期 (Shi et al., 2024; Zhang, L. et al., 2024),HBsAg 在这个过程中起着关键作用。Satoh 等人的开创性工作首次从生物化学角度将 HBsAg 定性为富含磷脂酰胆碱的颗粒 (Satoh et al., 1990),确立了 HBV 包膜的基本脂质组成。在此基础上,Núñez 及其同事揭示了 preS 结构域通过插入宿主磷脂双分子层介导膜融合的结构机制 (Núñez et al., 2009)。这些分子见解与临床观察相关,显示出 HBsAg 阳性患者血清磷脂谱的特征性改变,包括升高的磷脂酰胆碱 (PC)、
磷脂酰乙醇胺 (PE) 和溶血磷脂酸 (LPA),同时磷脂酰丝氨酸 (PS)、磷脂酰甘油 (PG)、磷脂酰肌醇 (PI) 和鞘磷脂降低(图 2 vi)(Huang, Q. et al., 2019)。宿主通过多种防御机制对抗 HBV 的脂质操纵。Sac1,一种关键的磷脂酰肌醇磷酸酶,通过以下方式破坏病毒传播:(i) 调节鞘脂合成以抑制 HBV 包膜向内体的运输 (Popescu et al., 2022),和 (ii) 促进自噬体-溶酶体融合以增强病毒颗粒的降解 (Zheng, J. et al., 2023)。此外,HBx 的 68-117 区域诱导的线粒体通透性改变——HBV 发病机制中的关键步骤——依赖于心磷脂重塑,而心磷脂重塑可能受铜蓝蛋白调节 (You et al., 2019)。
载脂蛋白劫持策略
最近的研究发现载脂蛋白,特别是载脂蛋白 E (ApoE),是 HBV 感染的关键介质。结构和功能研究表明,ApoE 被整合到 HBV 包膜中,在病毒发病机制中扮演三个关键角色(图 2 vii):(1) 通过稳定包膜结构促进病毒颗粒产生 (Qiao and Luo, 2019),(2) 通过与低密度脂蛋白受体 (LDLR) 相互作用介导病毒进入 (Li, Yingying and Luo, 2021),以及 (3) 通过遗传多态性影响宿主易感性 (Yin et al., 2010)。临床相关性表明,血清 ApoE 水平与慢性 HBV 进展密切相关 (Shen, Yueshuang et al., 2015),突显了其作为预后标志物和治疗靶点的潜力。HBV 还操纵其他载脂蛋白:载脂蛋白 H (ApoH) 特异性结合 HBsAg 以支持病毒持续存在 (Stefas, 2001),而 SHBs 下调 ApoAII,改变胆固醇谱 (Wu, Y. et al., 2024)。
最近的证据还表明 HBeAg 依赖性地调节其他载脂蛋白。CHB 患者血清载脂蛋白 M (ApoM) 水平升高,并且在 HBeAg 阴性个体中与 HBV DNA 载量呈正相关 (Shen, T. et al., 2016),提示 ApoM 在 HBV 相关脂质代谢和免疫调节中的作用。类似地,在 CHB 患者中观察到 ApoA1、ApoB、ApoC3 和 ApoA5 降低,并且在 HBeAg 阴性个体中,ApoC3 水平与 HBV DNA 载量呈负相关 (Cui, Y. et al., 2019)。这些在 HBeAg 阴性患者中的特异性关联尤其值得注意,因为这个阶段,虽然通常与低复制和最小炎症相关,但也可能包括由病毒变异驱动的活动性疾病个体。这些 HBeAg 依赖性关联突显了 HBV 更广泛的策略,即微调载脂蛋白网络,不仅是为了优化病毒感染性和逃避免疫反应,也是为了以基因型/复制状态依赖的方式调节宿主脂质代谢。
这些对载脂蛋白网络的协调修饰使 HBV 能够优化病毒感染性、逃避免疫反应并重编程肝脏脂质代谢。载脂蛋白在 HBV 生命周期中的关键参与提示了有前景的治疗策略,包括靶向 ApoE-LDLR 轴和开发载脂蛋白特异性调节剂。
代谢-临床交叉点:糖尿病-HBV 协同作用与激素调控网络
肝脏在维持全身葡萄糖平衡中起着核心作用,引出一个关键问题:HBV 感染是否会扰乱血糖调节?新出现的证据证实,慢性 HBV 感染通过代谢重编程和肝损伤破坏血糖控制,导致受影响的个体中空腹血糖受损 (IFG) 和糖尿病 (DM) 的患病率升高 (Huang, S.C. and Kao, 2024; Abu Baker et al., 2025)。DM 的风险与 HBV 相关肝病的严重程度相关,表现出层次性的患病率:HBV 相关肝硬化(风险最高)、慢性 HBV 感染(中等风险)、HBV 携带者(风险最低)(Shen, Y. et al., 2017)。值得注意的是,感染持续时间长和高病毒载量是 DM 发生的独立风险因素 (Shen, Yi et al., 2015)。在患有糖尿病的 HBV 感染者中,血糖控制往往不足。尽管如此,联合抗病毒和抗糖尿病治疗可能会改善结果 (Liu, Q. et al., 2025)。这些发现强调了在 CHB 患者中进行定期血糖异常筛查——尤其是口服葡萄糖耐量试验 (OGTT)——的重要性 (Mavrogiannaki et al., 2009),因为严格的血糖控制可能会改善该人群的临床结局 (Mak et al., 2023)。
HBV 干扰关键的葡萄糖调节激素,在病毒活性和代谢失调之间创造出双向相互作用。HBx 与胰岛素抵抗的发生有关,并似乎调节胰岛素信号通路 (Kim, H. Y. et al., 2012)。HBV 感染上调细胞内胰岛素受体,同时下调功能性细胞表面受体,损害胰岛素结合并加剧外周胰岛素抵抗。这种受体重新分布提高了循环葡萄糖水平,营造了促糖尿病环境 (Barthel et al., 2016)。另一方面,SHBs 激活胰高血糖素,上调肝脏糖异生,进一步破坏葡萄糖稳态 (Chen, Y. et al., 2022)。除了这两种激素,糖皮质激素 (GCs) 进一步放大了 HBV 感染中的代谢失调。GCs 是促进肝脏糖异生的类固醇激素 (Kuo et al., 2015)。值得注意的是,GCs 已被证明会增加类风湿性关节炎、哮喘和慢性阻塞性肺疾病 (COPD) 患者长期使用导致 HBV 再激活的风险 (Kim, T.W. et al., 2010; Chen, M.H. et al., 2017)。这种双重作用——既作为代谢干扰物又作为病毒再激活触发因素——突显了 HBV、GCs 和宿主代谢之间复杂的相互作用。当与干扰素联合使用时,GCs 通过恢复信号转导与转录激活因子的甲基化来恢复干扰素的抗病毒效力 (Bing et al., 2014)。这突显了 GCs 在 HBV 管理中的环境依赖性作用。
HBV 感染建立了代谢失调和肝功能障碍的恶性循环,驱动血糖异常并加速疾病进展。破译激素-病毒-代谢轴为改善 CHB 患者的病毒学和代谢结果提供了新途径。
结论与展望
HBV 感染与宿主细胞代谢之间的相互作用为治疗干预提供了一个有前景的途径。越来越多的证据强调了基因型对代谢通路的特异性调节,特别是通过 HBx 和聚合酶 rt269 多态性 (Lee et al., 2019; Choi et al., 2023; Schollmeier et al., 2024; Zhang, M. et al., 2025)。HBx 和聚合酶 rt269 多态性的变异塑造了不同的代谢结果:rt269I 诱导软骨样应激和 IFN-I 信号,而 rt269L 促进 HBx 稳定性和自噬。类似地,HBV 亚型 A1 和 A2 因影响蛋白质稳定性和结构的 HBx 多态性而在复制上存在差异。这些基因型依赖性特征影响线粒体自噬、ROS 产生和线粒体完整性,导致不同的临床结局。除了基因型特异性效应外,HBV 操纵关键的代谢途径,如糖酵解和 FA 氧化,以利于其复制和持续存在。靶向病毒诱导的代谢改变——例如抑制己糖激酶或乙酰辅酶 A 羧化酶——可以破坏 HBV 复制并降低病毒载量。此外,调节细胞能量调节因子,如 AMPK,不仅限制病毒传播,还能放大宿主细胞固有的抗病毒防御。值得注意的是,胆固醇代谢构成了 HBV 生命周期中的另一个关键弱点,因为该病毒利用宿主膜中富含胆固醇的微域进行病毒粒子组装、进入和分泌。针对胆固醇稳态的治疗策略——例如消耗质膜胆固醇或调节胆固醇衍生物(例如,氧化甾醇)——可能破坏 HBV 的结构完整性并削弱其感染性。这些代谢干预措施提供双重益处:直接的抗病毒作用和改善感染肝细胞的代谢适应性,同时解决病毒负担和相关的代谢失调。
尽管有这些见解,当前的 HBV 治疗主要依赖干扰素和核苷类似物。靶向代谢以增强治疗是未来研究的一个有前景的方向。代谢靶向虽然有希望,但受到机制理解差距的阻碍,这在很大程度上归因于现有模型的局限性。首先,传统的体外系统缺乏天然肝组织的结构和细胞复杂性,特别是在模拟肝细胞与非实质细胞之间的动态相互作用方面。其次,虽然人源化嵌合小鼠代表了细胞培养系统的进步,但它们不完全再现自然感染中观察到的人类病毒动力学和免疫反应的完整谱系。这些局限性需要谨慎解释当前的发现,并强调需要解决几个关键的未解问题,包括:(i) 慢性 HBV 感染与 MASLD 之间明显的保护性关联的潜在机制是什么——HBV 是直接抑制脂肪生成途径还是通过免疫调节间接作用?(ii) 病毒诱导的代谢重编程(例如,增强的糖酵解,减少的 β-氧化)在多大程度上驱动肝病进展,如纤维化或 HCC?(iii) 系统性代谢紊乱(例如,糖尿病、肥胖)如何与 HBV 感染相互作用并影响病毒发病机制或治疗反应?(iv) 营养或饮食干预(例如,生酮或间歇性禁食)能否调节 HBV 诱导的代谢变化,从而发挥抗病毒益处?(v) 在 HBV 感染期间,非实质肝脏细胞对代谢反应的贡献是什么,细胞间代谢的相互作用如何影响病毒持续存在?
为应对这些挑战,可以探索几个有前景的研究方向:(1) 包含肝星状细胞、库普弗细胞和肝窦内皮细胞的先进 3D 共培养平台可能更好地模拟肝脏的代谢微环境;(2) 具有重建适应性免疫的下一代人源化模型可以为研究免疫-代谢-病毒相互作用提供更生理相关的平台;以及 (3) 结合多组学方法与详细代谢表型的纵向临床研究可能有助于弥合实验发现与人类病理生理学之间的差距。这种整合方法可以更细致地理解 HBV 感染如何与宿主代谢途径相交,可能在这个关键界面揭示新的治疗靶点。
参考文献:Yan Y, Wei Z, Zheng M, Lu M, Wang X. HBV and host metabolic crosstalk: Reprogramming pathways for viral replication and pathogenesis. Virol Sin. 2025 Oct;40(5):685-693. doi: 10.1016/j.virs.2025.09.008.
https://pmc.ncbi.nlm.nih.gov/articles/PMC12665413/
原文链接:https://doi.org/10.1016/j.virs.2025.09.008