编者按
我们将推出一个新的栏目,重构算法的介绍,在这个栏目中,我们将介绍omicverse开源重构生态最新的成果以及完整的教程,欢迎关注。
本期教程里,我们给大家带来的是Bulk TCR免疫组库分析:这是omicverse中 ov.airr 模块的内容。我们在omicverse中整合了bulk immune repertoire分析常用的一套流程,包括克隆型统计、CDR3长度分布、克隆扩增、多样性、公共克隆型、V基因使用、CDR3 motif以及VDJdb抗原注释。
本文使用一个多发性硬化症(MS)患者和健康对照的TCR beta链数据,演示如何用OmicVerse完成一套完整的bulk TCR免疫组库分析,希望本期教程对你有所帮助。
本文AI率10%,教程较长,全文阅读大约需要20min。
OmicVerse团队
1. 引言
免疫组库分析里,TCR其实是一类很特殊的数据。
它不像单细胞RNA-seq那样是一张“细胞 × 基因”的表达矩阵,而更像是一张克隆型表。每一行通常包含一个CDR3序列、V/J基因、出现次数,以及这个克隆型来自哪个样本。
所以对于bulk TCR分析,我们一般需要解决的问题为:
这些问题看起来都很基础,但它们构成了bulk TCR分析的主干。
在实际项目里,如果你拿到的是疫苗前后、治疗前后,或者疾病与健康对照的bulk TCR数据,如果你把omicverse里的分析都跑一遍,出一遍图。那么你就自然而然能看见,样本之间的差异,包括克隆扩增、多样性或者V基因使用等内容。
本期教程使用的是 pyimmunarch 里的示例数据,一共12个TCR beta链样本,其中6个来自MS患者,6个来自健康对照。
2. Bulk TCR能分析什么
在OmicVerse里,bulk TCR分析主要通过 ov.airr 完成。
| | |
|---|
| repertoire_summary | |
| spectratype_bulk | |
| clonality | |
| repertoire_diversity | |
| repertoire_overlap_bulk | |
| public_clonotypes | |
| gene_usage_bulk | |
| track_clonotypes | |
| kmer_analysis | |
| annotate_antigen_bulk | |
omicverse中,我们把免疫组库拆成了几个层面:数量、多样性、重叠、序列模式和抗原信息。
3. 数据读入
先载入OmicVerse并设置绘图风格:
import omicverse as ov
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
ov.plot_set()
读取bulk TCR示例数据:
immdata = ov.airr.load_example_immdata()
print(f"samples: {len(immdata.data)}")
print(f"sample names: {list(immdata.data)}")
immdata.meta
samples: 12
sample names: ['A2-i129', 'A2-i131', 'A2-i132', 'A2-i133', 'A4-i191', 'A4-i192',
'MS1', 'MS2', 'MS3', 'MS4', 'MS5', 'MS6']
这里一共有12个样本。前6个是健康对照,后6个是MS患者。
cohort = ov.airr.cohort_groups(
immdata,
status_col="Status",
mapping={"MS": "MS", "C": "Healthy"},
)
meta = cohort["meta"]
hc_samples = cohort["samples"]["Healthy"]
ms_samples = cohort["samples"]["MS"]
print(f"MS patients ({len(ms_samples)}): {ms_samples}")
print(f"healthy ({len(hc_samples)}): {hc_samples}")
MS patients (6): ['MS1', 'MS2', 'MS3', 'MS4', 'MS5', 'MS6']
healthy (6): ['A2-i129', 'A2-i131', 'A2-i133', 'A2-i132', 'A4-i191', 'A4-i192']
这一步其实就是把样本分组整理好。后面所有比较都会围绕 MS 和 Healthy 两组展开。
4. 样本克隆型分布
对于Bulk TCR分析,我们先看每个样本的unique clonotypes。
explore = ov.airr.repertoire_summary(immdata, meta=meta)
explore
unique_clonotypes total_clones group
A2-i129 6532 8500 Healthy
A2-i131 6553 8500 Healthy
A2-i132 6849 8500 Healthy
A2-i133 6393 8500 Healthy
A4-i191 5146 8500 Healthy
A4-i192 5823 8500 Healthy
MS1 5405 8500 MS
MS2 7145 8500 MS
MS3 6461 8500 MS
MS4 7447 8500 MS
MS5 5657 8500 MS
MS6 7409 8500 MS
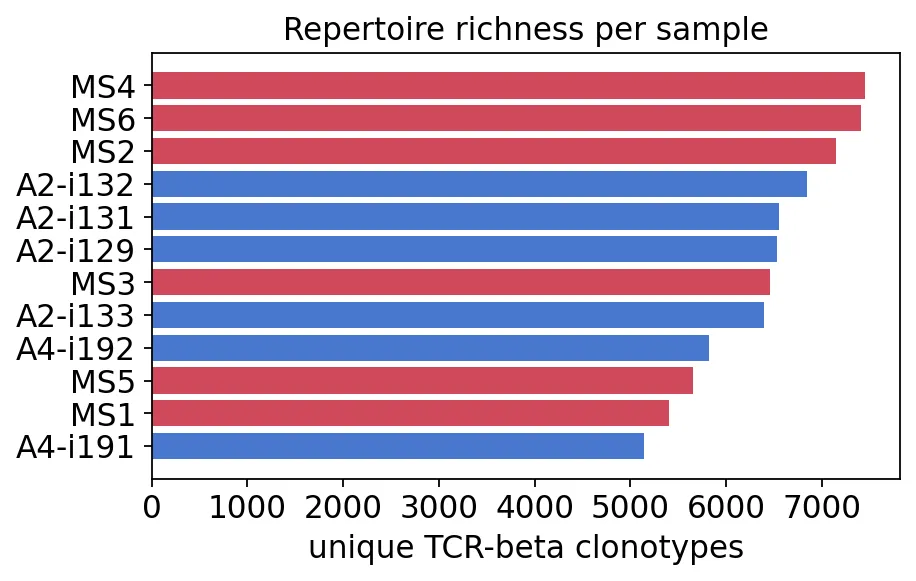 每个样本的unique clonotypes
每个样本的unique clonotypes这个数据里,每个样本都抽到了8500个clones,但unique clonotypes数量不同。
MS组并不是简单地“克隆型更少”。比如MS4和MS6的unique clonotypes反而比较高。这一点在后面多样性分析里还会看到。
5. CDR3长度分布
接下来可以看CDR3长度分布,也就是免疫组库分析里常说的spectratype。
spec = ov.airr.spectratype_bulk(immdata, groupby=cohort["groups"])
spec_ms, spec_hc = spec["MS"].dropna(), spec["Healthy"].dropna()
print(f"CDR3-length spectratype: MS peak {spec_ms.idxmax()} aa, healthy peak {spec_hc.idxmax()} aa")
CDR3-length spectratype: MS peak 15 aa, healthy peak 14 aa
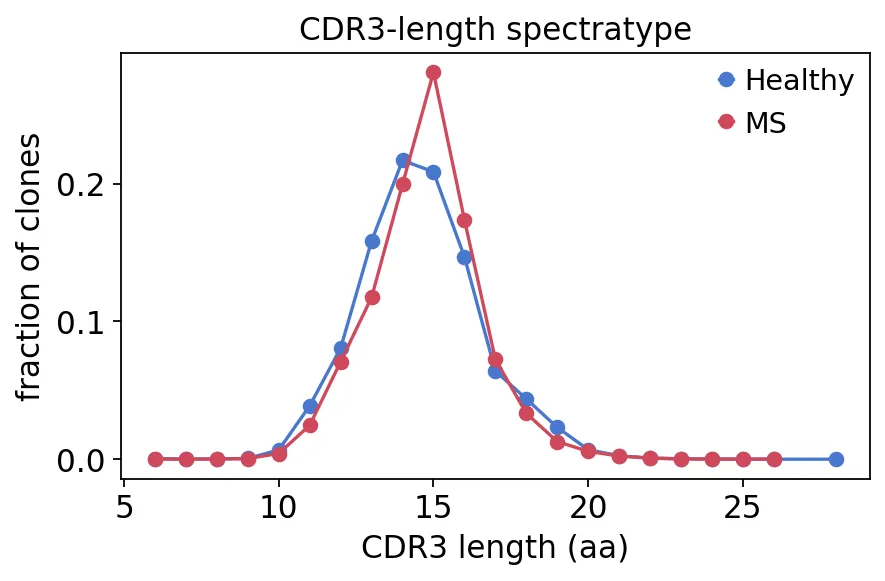 CDR3长度分布
CDR3长度分布可以看到,MS组的CDR3长度峰值在15 aa,健康对照峰值在14 aa。
这不意味着MS一定会导致CDR3长度增加。更准确地说,在这个示例队列里,MS组和健康组的CDR3长度分布有一个轻微偏移。具体的偏移背后的内容,还要结合样本量和统计检验去看。
6. 克隆扩增
在TCR分析中,有一个很重要的问题:这个样本是不是被少数克隆占据了?
如果前10个克隆型就占了很高比例,那么说明这个样本里可能存在明显的克隆扩增。
top_clones = ov.airr.clonality(immdata, method="top")
top10 = top_clones.iloc[:, 0].rename("top10_proportion").to_frame()
ov.airr.summarize_by_group(
top10,
group=meta.set_index("Sample")["group"],
metrics=("mean", "min", "max"),
)
top10_proportion
mean min max
group
Healthy 0.093824 0.023882 0.171765
MS 0.109804 0.023294 0.206118
我们发现,在MS组top10克隆占比的均值是0.1098,健康对照是0.0938。这个差距不大,不过MS组最高样本能到0.2061,说明个别MS样本里有更强的克隆扩增。
再看不同扩增等级的比例:
homeo = ov.airr.clonality(immdata, method="homeo")
homeo_by_group = ov.airr.summarize_by_group(
homeo,
group=meta.set_index("Sample")["group"],
metrics=("mean",),
)
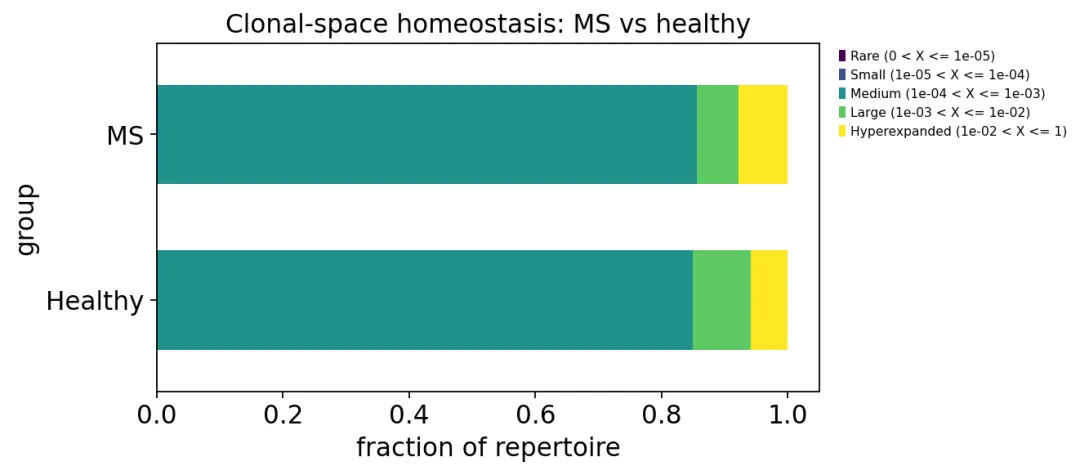 克隆扩增等级
克隆扩增等级这里可以看到MS组的hyperexpanded比例略高,健康对照的large clone比例略高。这个结果比较符合我们前面对top10比例的观察:MS组不是所有样本都被克隆扩增支配,只是有几个样本的扩增更突出。
7. 多样性分析
免疫组库多样性不能只看unique clonotypes。因为一个样本可能有很多不同TCR,但其中一部分TCR只有很低丰度;另一个样本可能unique clonotypes少一点,但分布更均匀。
这里先计算Chao1和D50。
chao1 = ov.airr.repertoire_diversity(immdata, method="chao1")
chao1 = chao1.join(meta.set_index("Sample")[["group"]])
ov.airr.summarize_by_group(chao1, group="group", metrics=("mean", "std"))
Estimator
mean std
group
Healthy 40689.395042 7999.299275
MS 68678.298416 21989.281690
d50 = ov.airr.repertoire_diversity(immdata, method="d50")
d50 = d50.join(meta.set_index("Sample")[["group"]])
ov.airr.summarize_by_group(d50, group="group", metrics=("mean", "min", "max"))
Clones
mean min max
group
Healthy 1878.666667 861.0 2393.0
MS 2305.500000 1111.0 3159.0
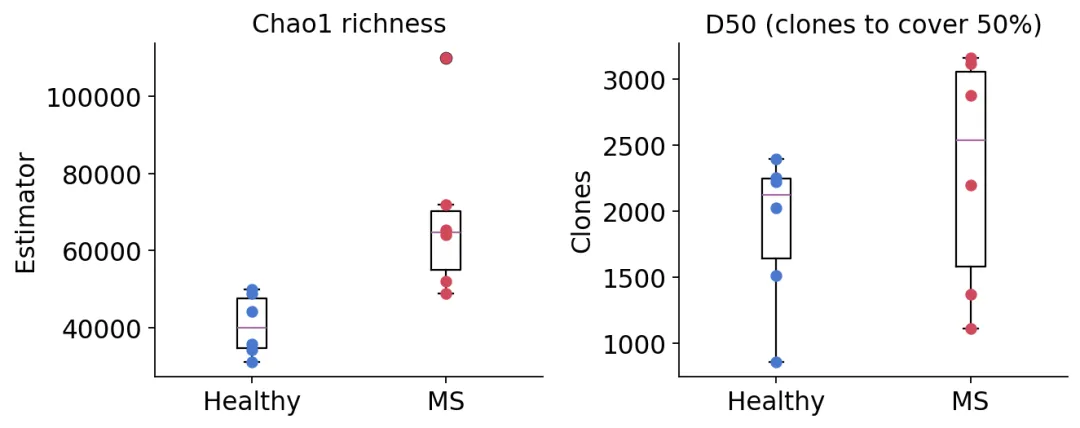 Chao1和D50多样性
Chao1和D50多样性在这个数据中,MS组的Chao1和D50均值都更高。
这可能会有点反直觉。很多人会下意识觉得疾病状态应该对应“多样性下降”或者“克隆扩增增强”。但在这个示例数据里,MS组更像是存在更高的潜在richness,同时部分样本又有克隆扩增。
8. Hill number和稀释曲线
Hill number可以理解成一组带有不同权重的多样性指标。
当Q比较低时,它更重视稀有克隆;当Q比较高时,它更重视高丰度克隆。
hill = ov.airr.repertoire_diversity(immdata, method="hill")
hill = hill.merge(meta[["Sample", "group"]], on="Sample")
hill_profile = ov.airr.summarize_by_group(hill, group="group", value="Diversity")
hill_profile
group Healthy MS
Q
1 3864.967000 4264.817350
2 1107.316904 1758.151055
3 467.964705 723.051209
4 285.296948 414.225282
5 211.362082 301.823245
6 173.527488 247.319992
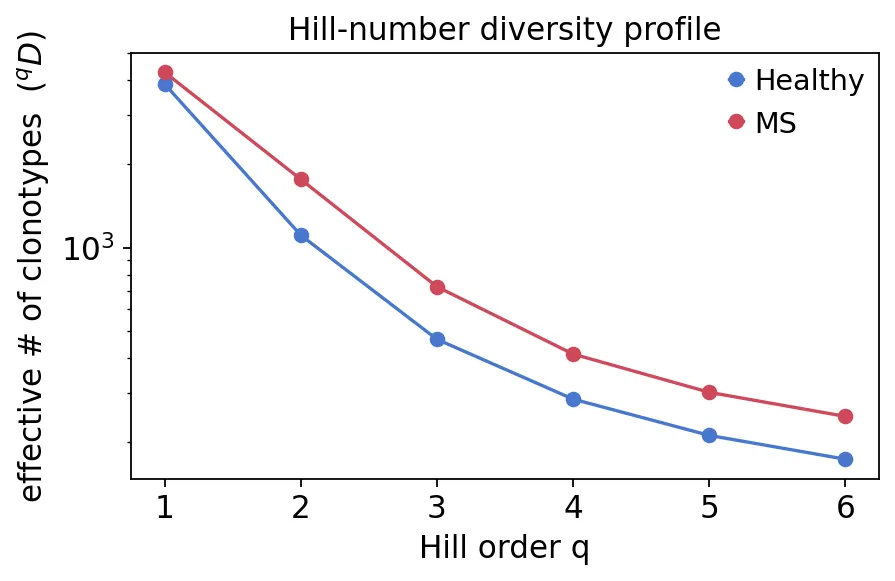 Hill number diversity profile
Hill number diversity profile从Q1到Q6,MS组都高于健康对照。
再看稀释曲线:
raref = ov.airr.repertoire_diversity(immdata, method="raref")
raref = raref.merge(meta[["Sample", "group"]], on="Sample")
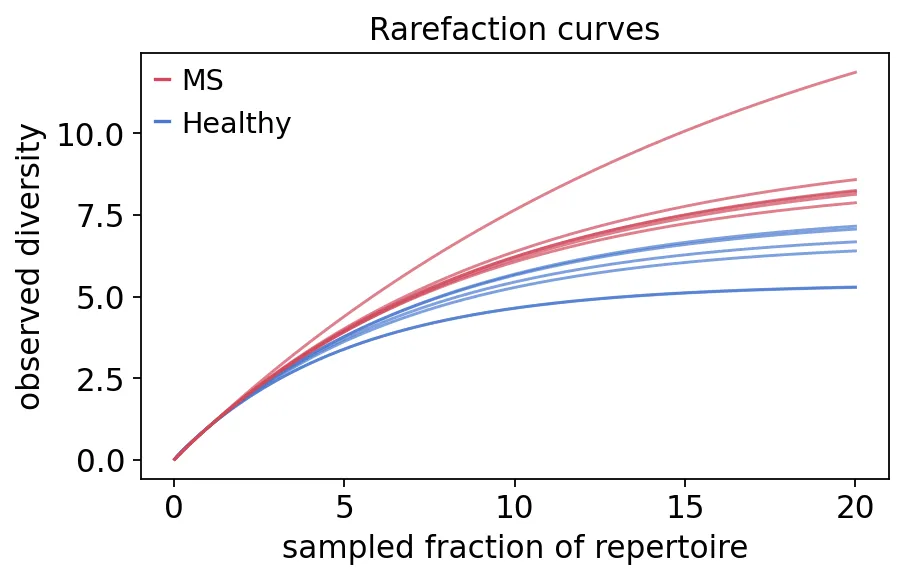 稀释曲线
稀释曲线稀释曲线的用途很直接:它告诉我们在不同抽样深度下,能观察到多少克隆型。
在真实项目里,如果两组测序深度差异很大,那么这张图就能有效避免我们直接比较unique clonotypes然后被测序深度带偏。
9. 共享和公共克隆型
接下来分析样本之间共享多少TCR克隆型。
overlap = ov.airr.repertoire_overlap_bulk(immdata, method="public")
overlap = overlap.loc[hc_samples + ms_samples, hc_samples + ms_samples]
overlap.iloc[:5, :5]
A2-i129 A2-i131 A2-i133 A2-i132 A4-i191
A2-i129 NaN 63.0 74.0 69.0 46.0
A2-i131 63.0 NaN 56.0 81.0 42.0
A2-i133 74.0 56.0 NaN 87.0 49.0
A2-i132 69.0 81.0 87.0 NaN 62.0
A4-i191 46.0 42.0 49.0 62.0 NaN
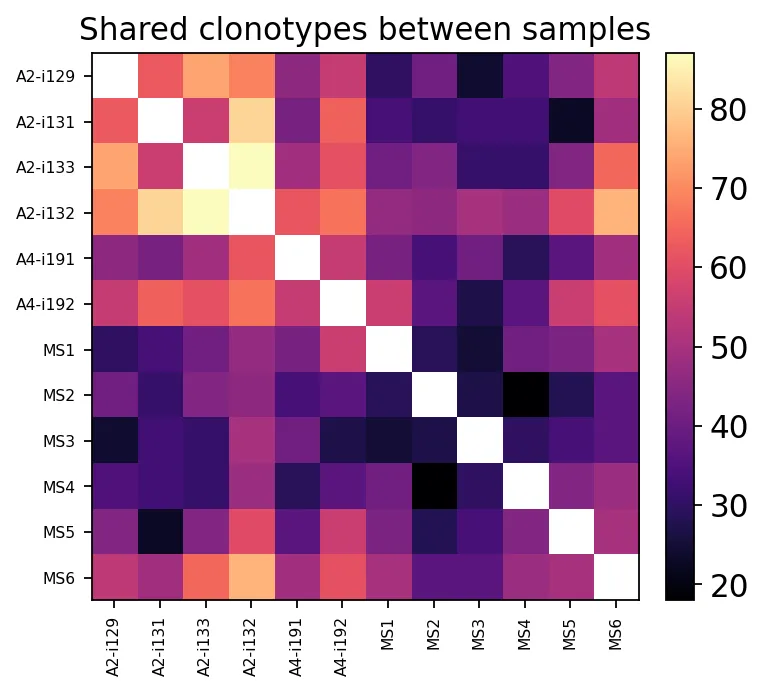 样本间共享克隆型热图
样本间共享克隆型热图在热图中,我们能看见样本两两之间共享克隆型的数量。
但直接共享数量会受到样本深度和克隆丰度影响,因此我们也可以看Morisita-Horn这类更稳一些的重叠指标:
mh = ov.airr.repertoire_overlap_bulk(immdata, method='morisita')
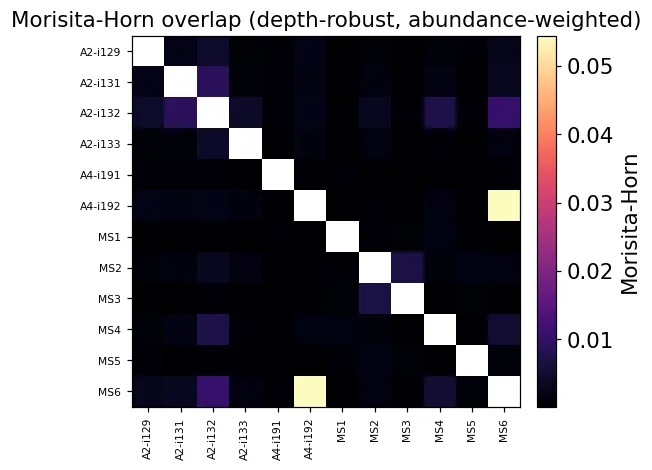 Morisita-Horn重叠热图
Morisita-Horn重叠热图然后统计所有公共克隆型:
pubrep = ov.airr.public_clonotypes(immdata, col="aa+v")
shared = pubrep["Samples"].value_counts().sort_index()
print(f"public-repertoire table: {pubrep.shape[0]} clonotypes")
print(f"clonotypes in all 12 samples: {(pubrep['Samples'] == 12).sum()}")
shared
public-repertoire table: 74444 clonotypes
clonotypes in all 12 samples: 0
Samples
1 73933
2 415
3 66
4 18
5 8
6 3
8 1
Name: count, dtype: int64
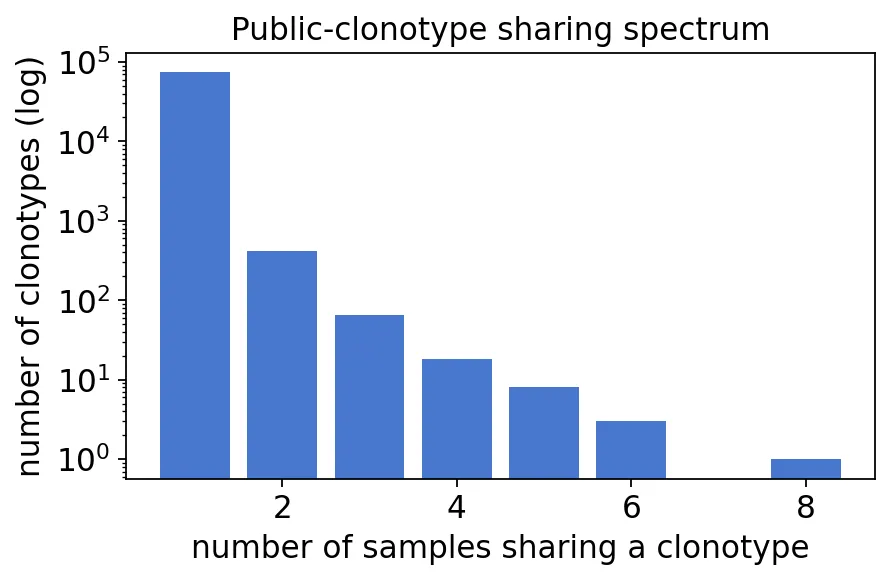 公共克隆型共享样本数
公共克隆型共享样本数我们发现,绝大多数TCR克隆型只出现在一个样本里,真正跨多个样本重复出现的克隆型很少。
这也是为什么TCR公共克隆型分析要很谨慎。一个克隆型在多个样本出现,可能是共同抗原刺激,也可能是生成概率高的public TCR,还可能是数据库或测序噪音带来的影响。
10. V基因使用偏好
除了CDR3序列本身,我们还可以看TRBV基因的使用比例。
gene_usage = ov.airr.gene_usage_bulk(immdata, gene="hs.trbv", norm=True)
gu_diff = ov.airr.differential_gene_usage(
gene_usage,
groups=cohort["groups"],
group1="MS",
group2="Healthy",
)
gu_diff.reindex(gu_diff["delta"].abs().sort_values().index[-8:])
Healthy MS delta
Names
TRBV4-3 0.028401 0.011899 -0.016502
TRBV9 0.035604 0.022978 -0.012626
TRBV2 0.027394 0.017819 -0.009575
TRBV7-2 0.060213 0.051772 -0.008441
TRBV4-1 0.032536 0.041145 0.008609
TRBV6-5 0.028422 0.038060 0.009638
TRBV12-4 0.072484 0.089986 0.017502
TRBV5-1 0.065543 0.100274 0.034731
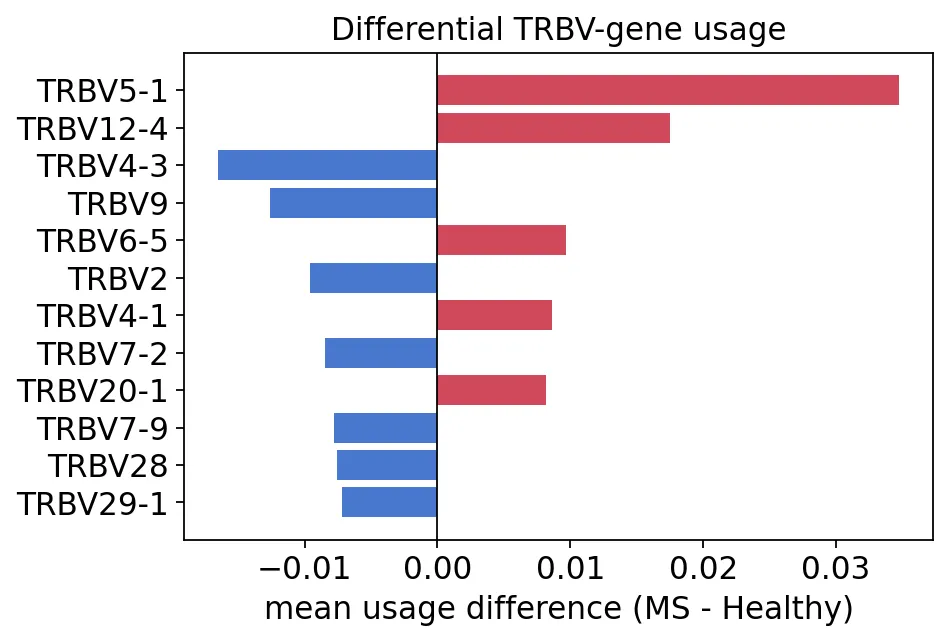 TRBV基因使用差异
TRBV基因使用差异在这个数据里,MS组TRBV5-1和TRBV12-4使用比例更高,健康对照组TRBV4-3、TRBV9、TRBV2更高。
我觉得V基因使用差异适合作为一个辅助结果。它不一定能直接解释疾病机制,但能告诉我们两组样本的TCR生成或选择过程可能有偏移。
11. 克隆追踪
如果我们想知道某个样本的top clone是否也出现在其他样本里,可以用 track_clonotypes。
这里取MS1中丰度最高的10个CDR3:
track = ov.airr.track_clonotypes(immdata, which=("MS1", 10), col="aa")
track_mat = track.set_index("CDR3.aa")[hc_samples + ms_samples]
track_mat
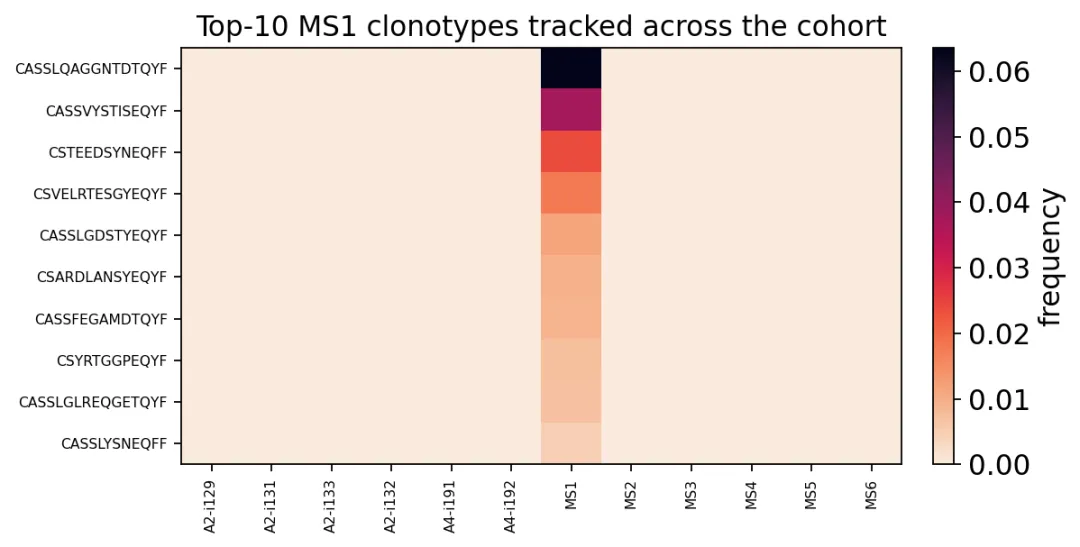 MS1 top clones跨样本追踪
MS1 top clones跨样本追踪我们发现,MS1里的top clone基本上还是以MS1为主,没有形成特别明显的跨样本共享。
在纵向随访项目里,我们可以通过这个图,来分析例如治疗前出现的克隆,在治疗后是否消失;疫苗接种后扩增的克隆,几个月后是否还存在。
12. CDR3 k-mer和motif
TCR CDR3序列不是随机字符串。
如果某些氨基酸片段反复出现,说明这个队列里可能存在偏好的序列模式。我们可以先做5-mer统计:
kmers = ov.airr.kmer_analysis(immdata, k=5, col="aa")
kmer_total = kmers.set_index("Kmer").sum(axis=1).sort_values(ascending=False)
print(f"distinct 5-mers: {len(kmer_total):,} across 12 samples")
kmer_total.head(10)
distinct 5-mers: 172,926 across 12 samples
Kmer
CASSL 13796.0
NEQFF 11897.0
YEQYF 11370.0
ETQYF 10571.0
DTQYF 8535.0
YNEQF 6979.0
CASSP 6391.0
CASSQ 6204.0
TDTQY 5301.0
YGYTF 4386.0
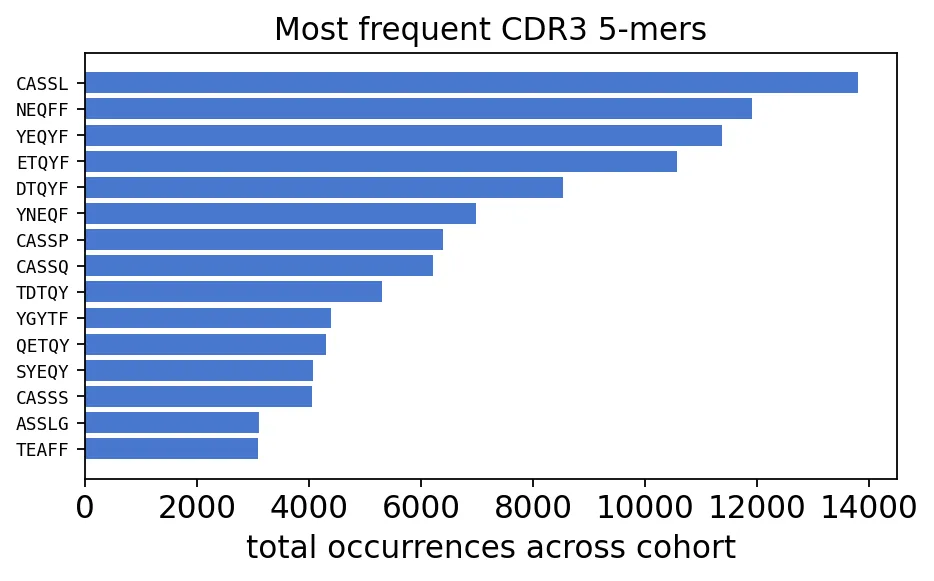 Top CDR3 5-mers
Top CDR3 5-mers这些top 5-mer很符合TCR beta链CDR3的常见结构。比如CASSL、CASSP、CASSQ通常来自CDR3起始区域,NEQFF、YEQYF、ETQYF则更接近J基因相关区域。
接着,我们可以把k-mer进一步压成motif:
pfm = ov.airr.kmer_motif(kmers, method="freq")
pwm = ov.airr.kmer_motif(kmers, method="wei")
print(f"motif profile: {pfm.shape[0]} amino acids x {pfm.shape[1]} positions")
print("dominant residue per position:", pfm.idxmax(axis=0).to_dict())
motif profile: 20 amino acids x 5 positions
dominant residue per position: {'V1': 'S', 'V2': 'S', 'V3': 'S', 'V4': 'G', 'V5': 'F'}
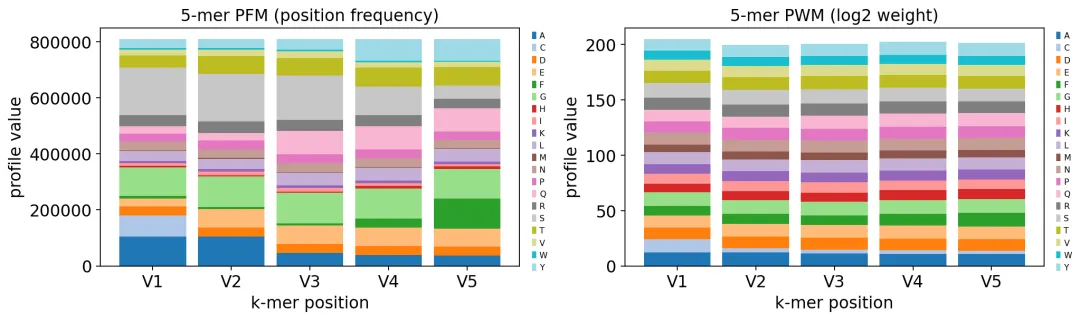 CDR3 5-mer motif
CDR3 5-mer motifmotif图不能直接告诉我们抗原是什么,但可以展示这个队列里CDR3序列的共同模式。
13. CDR3氨基酸理化性质
除了k-mer,我们还可以把CDR3转成氨基酸理化性质,比如疏水性、电荷和体积。
这里以MS1为例:
prop_table = ov.airr.cdr3_aa_properties(
immdata,
sample="MS1",
property=["hydropathy", "charge", "volume"],
max_len=18,
align="left",
)
print(f"MS1 CDR3 length used: {prop_table.shape[0]} positions")
prop_table.head()
MS1 CDR3 length used: 18 positions
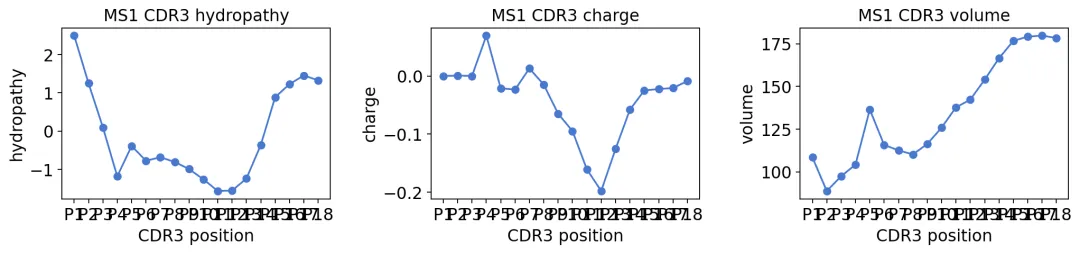 CDR3理化性质曲线
CDR3理化性质曲线还可以画氨基酸组成:
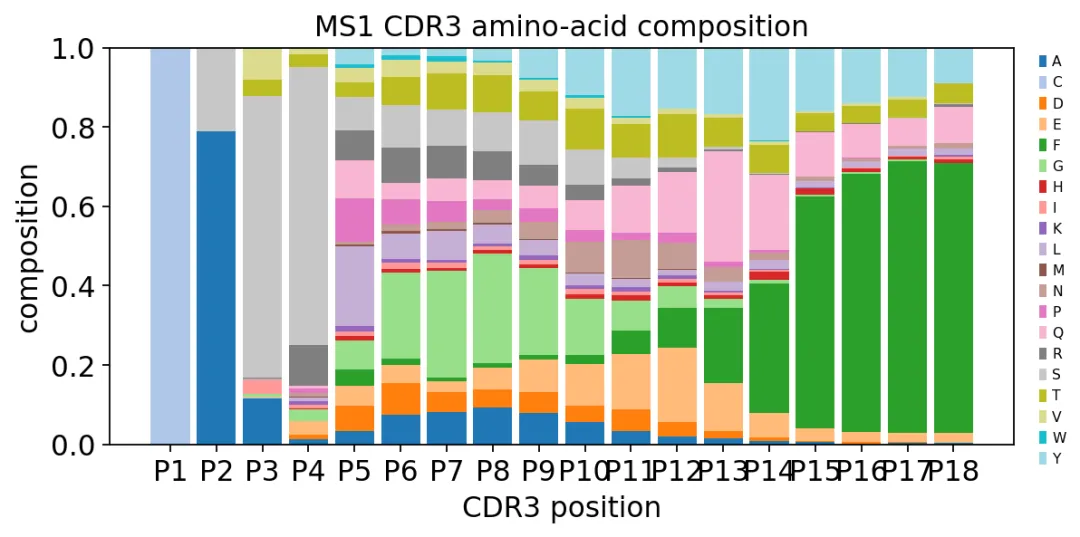 CDR3氨基酸组成
CDR3氨基酸组成再比较MS和健康对照的疏水性:
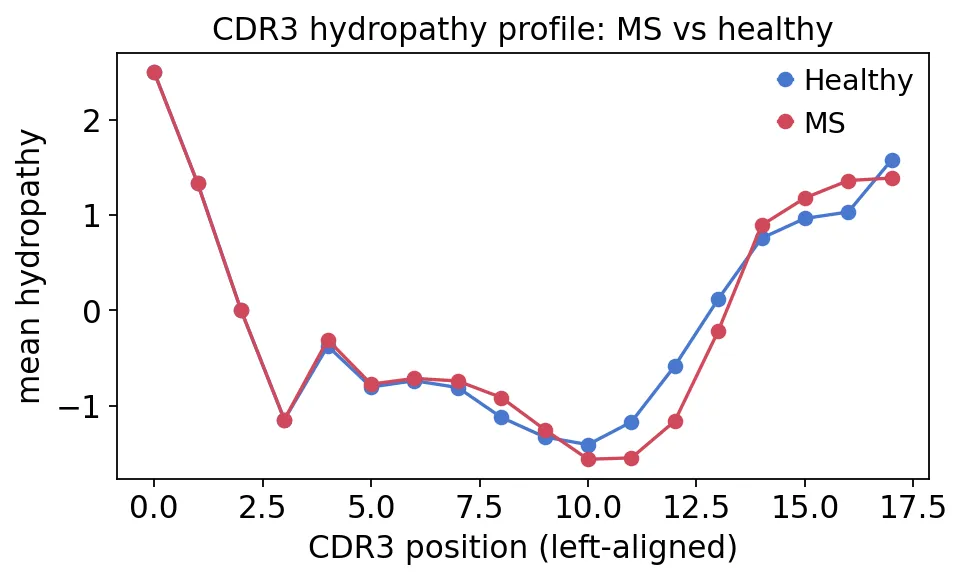 MS与健康对照CDR3疏水性比较
MS与健康对照CDR3疏水性比较上述这些图可以有助于帮助我们描述序列层面的整体偏好。如果某个疾病队列里CDR3理化性质出现很稳定的偏移,那么就值得进一步分析讨论
14. 多变量V基因使用分析
前面我们看的是单个TRBV基因的差异。现在可以把所有TRBV基因的使用比例放在一起,看样本在整体V基因空间里的距离。
groups = dict(zip(meta["Sample"], meta["group"]))
gu_res = ov.airr.gene_usage_analysis(
gene_usage,
distance="js",
reduction="mds",
clustering="hclust",
k=2,
groups=groups,
)
print("result keys:", list(gu_res.keys()))
gu_res["embedding"].head()
result keys: ['distance', 'embedding', 'clusters', 'kruskal']
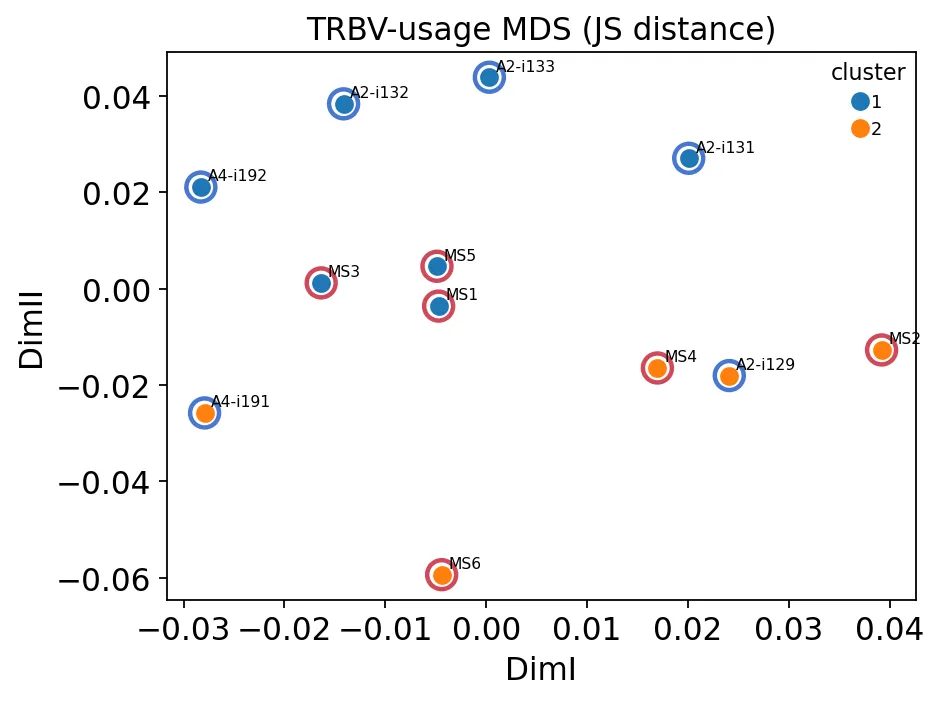 TRBV使用MDS
TRBV使用MDS可以看到,MS和健康对照并没有被非常干净地分成两团,而是有一定混合。
这其实很常见。免疫组库里的疾病信号很多时候没有一个“开关式”的分类边界,它更像是分散在多个指标里的弱偏移。
再看Kruskal检验结果:
kw = gu_res["kruskal"].dropna().head(10).copy()
kw["signif"] = np.where(kw["p_value"] < 0.05, "*", "")
print(f"genes tested: {gu_res['kruskal'].shape[0]}")
print(f"nominally significant (p<0.05): {(gu_res['kruskal']['p_value'] < 0.05).sum()}")
kw
genes tested: 48
nominally significant (p<0.05): 9
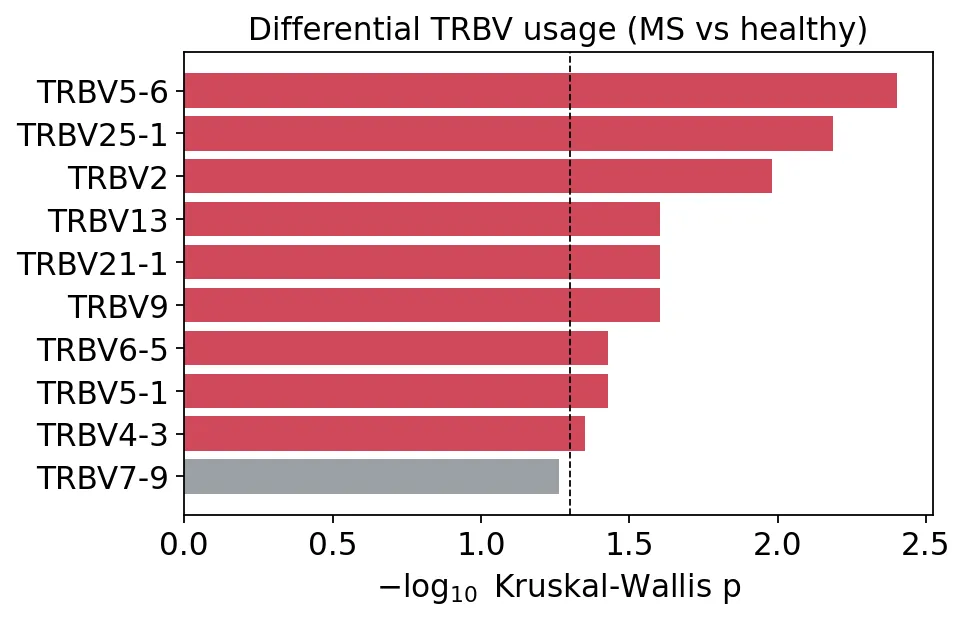 TRBV基因Kruskal检验
TRBV基因Kruskal检验这里有9个TRBV基因在名义p值上小于0.05,包括TRBV5-6、TRBV25-1、TRBV2、TRBV13、TRBV21-1、TRBV9、TRBV6-5、TRBV5-1和TRBV4-3。
样本量只有12个,因此我们可以把它们当作候选信号,后续在更大队列里验证。
15. Repertoire overlap结构
类似地,我们也可以基于样本间overlap做MDS。
ov_res = ov.airr.overlap_analysis(
overlap.fillna(0.0),
method="mds+hclust",
k=2,
)
print("overlap-analysis keys:", list(ov_res.keys()))
overlap-analysis keys: ['coords', 'clusters']
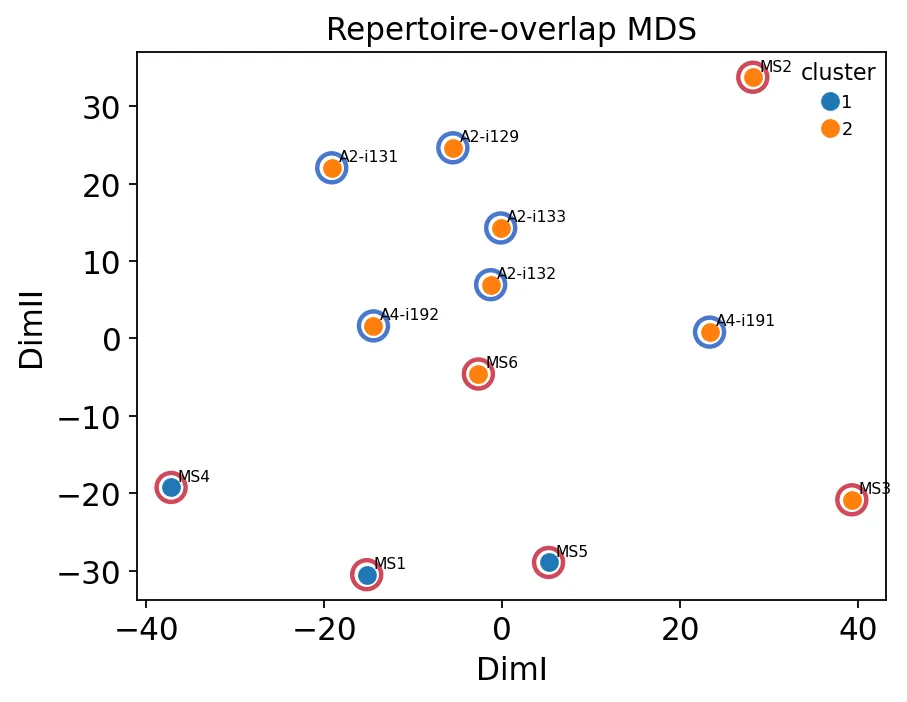 样本重叠结构MDS
样本重叠结构MDS再看分组和聚类的一致性:
cross = pd.crosstab(ov_emb["group"], ov_emb["Cluster"])
purity, agree = ov.airr.cluster_purity(
ov_emb["Cluster"],
ov_emb["group"],
return_assignment=True,
)
print("group x overlap-cluster contingency:")
print(cross)
print(f"best cluster-vs-group agreement: {purity:.0%}")
group x overlap-cluster contingency:
Cluster 1 2
group
Healthy 0 6
MS 3 3
best cluster-vs-group agreement: 75%
这个结果跟V基因使用MDS类似:健康对照相对更集中,而MS组内部异质性更强。
16. Public repertoire workflow
我们还可以把公共克隆型按组拆开。
pr_ms = ov.airr.public_repertoire(
immdata,
col="aa+v",
filter_by={"Status": "MS"},
)
pr_hc = ov.airr.public_repertoire(
immdata,
col="aa+v",
filter_by={"Status": "C"},
)
print(f"MS public repertoire : {pr_ms.shape[0]:,} clonotypes")
print(f"healthy public repertoire: {pr_hc.shape[0]:,} clonotypes")
MS public repertoire : 38,579 clonotypes
healthy public repertoire: 36,141 clonotypes
然后看MS内部共享情况:
ms_stats = ov.airr.public_repertoire(
immdata,
col="aa+v",
filter_by={"Status": "MS"},
statistics=True,
)
print(f"MS-shared clonotype groups: {ms_stats.shape[0]}")
print(f"top MS-internal sharing: {ms_stats.iloc[0]['Count']} clonotypes in pair {ms_stats.iloc[0]['Group']}")
ms_stats.head(8)
MS-shared clonotype groups: 26
top MS-internal sharing: 11 clonotypes in pair MS1&MS4
Group Count
0 MS1&MS4 11
1 MS4&MS5 11
2 MS4&MS6 9
3 MS1&MS5 9
4 MS5&MS6 9
5 MS2&MS3 8
6 MS3&MS6 8
7 MS1&MS3 7
最后比较MS和健康对照都出现的公共克隆型:
cmp = ov.airr.public_repertoire(
immdata,
col="aa+v",
filter_by={"Status": "MS"},
compare_to=pr_hc,
)
print(f"clonotypes public in BOTH MS and healthy: {cmp.shape[0]}")
clonotypes public in BOTH MS and healthy: 276
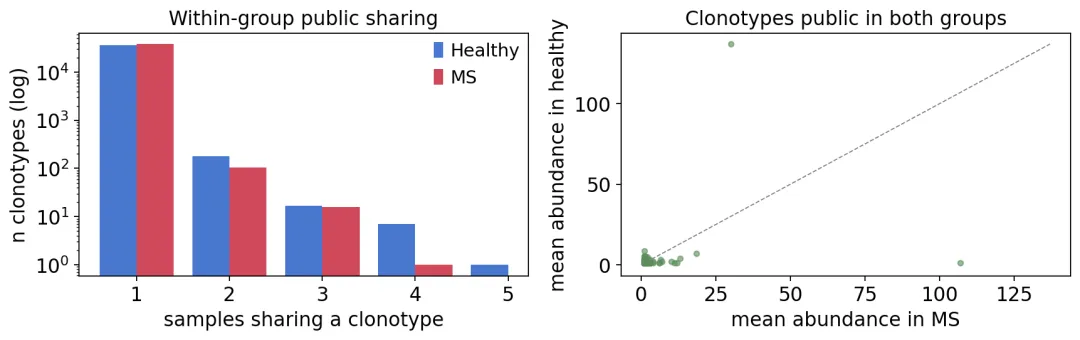 组内与组间公共克隆型比较
组内与组间公共克隆型比较这张图的意义在于把“公共克隆型”拆得更清楚。
有些克隆型只在MS内部共享,有些只在健康对照内部共享,还有276个克隆型同时出现在两组。后者通常不能直接解释成疾病特异TCR,因为它们也出现在健康人里。
17. VDJdb抗原注释
最后一步,我们把bulk TCR克隆型和VDJdb做匹配。
VDJdb是一个整理TCR和已知抗原关系的数据库。它不是万能的,命中率通常不会很高,但如果能匹配到CMV、EBV、Influenza这类常见抗原,可以给我们一个抗原来源层面的参考。
vdjdb = ov.datasets.vdjdb_reference()
print(f"real VDJdb: {vdjdb.shape[0]:,} TCR-epitope records, {vdjdb.shape[1]} columns")
print(vdjdb["gene"].value_counts().to_string())
vdjdb_trb = vdjdb[vdjdb["gene"] == "TRB"].copy()
print(f"TRB subset used for annotation: {vdjdb_trb.shape[0]:,} records")
real VDJdb: 132,204 TCR-epitope records, 10 columns
gene
TRB 87992
TRA 44212
TRB subset used for annotation: 87,992 records
然后进行匹配:
ann = ov.airr.annotate_antigen_bulk(
immdata,
db=vdjdb_trb,
data_col="CDR3.aa",
db_col="cdr3_aa",
)
print(
f"cohort clonotypes matching VDJdb: {ann.shape[0]:,} "
f"(of {ann['CDR3.aa'].nunique():,} unique CDR3.aa - a 2.1% hit rate)"
)
ann.head()
cohort clonotypes matching VDJdb: 1,615 (of 76,105 unique CDR3.aa - a 2.1% hit rate)
部分top匹配结果如下:
再按样本和抗原来源做一个总览:
sample_cols = hc_samples + ms_samples
load = ov.airr.antigen_load_summary(
ann,
sample_cols=sample_cols,
meta=meta,
)
load.groupby("group")["total_clones"].sum()
total VDJdb-annotated clones per group:
Healthy 1920.0
MS 1473.0
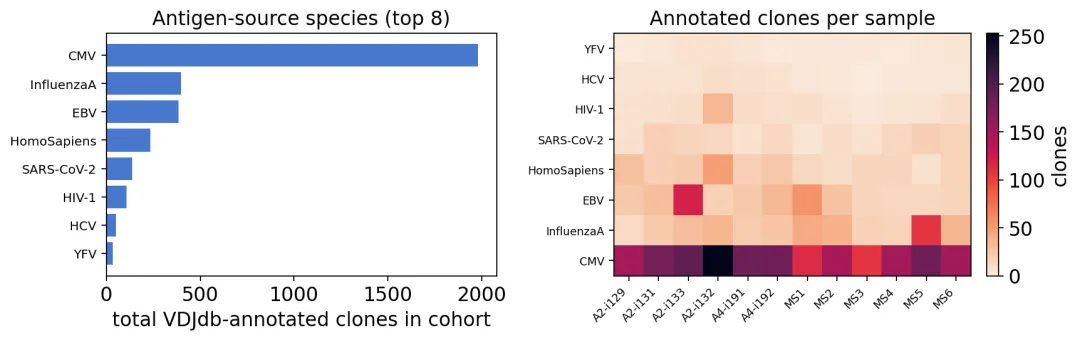 VDJdb抗原注释
VDJdb抗原注释可以看到,VDJdb能匹配到的抗原主要来自CMV、EBV和Influenza。
VDJdb匹配结果更多只是一个参考。因为2.1%的命中率有点低,其实在真实场景下,绝大多数TCR没有已知抗原注释。因此VDJdb的结果可以用来解释“这个队列里能否看到一些已知病毒相关TCR”,而不是直接给每个TCR贴上抗原标签。
18. 总结
在本期教程中,我们用12个bulk TCR beta链样本跑完了一套MS患者和健康对照的免疫组库分析流程:
- 这个队列包含6个MS样本和6个健康对照样本,每个样本抽取8500个clones。
- MS组CDR3长度峰值在15 aa,健康对照峰值在14 aa。
- MS组top10克隆占比均值为0.1098,健康对照为0.0938,提示MS组部分样本存在更强的克隆扩增。
- MS组Chao1和D50均值都高于健康对照,说明这个数据里MS并不是简单的多样性下降。
- 大多数TCR克隆型只出现在一个样本中,12个样本全部共享的克隆型为0。
- MS组TRBV5-1和TRBV12-4使用比例更高,健康对照组TRBV4-3、TRBV9、TRBV2更高。
- V基因使用和overlap MDS都显示MS与健康对照不是完全分开的两团,MS组内部异质性更明显。
- VDJdb匹配到1,615个已知抗原相关克隆型,命中率约2.1%,主要对应CMV、EBV和Influenza。
实际上我觉得bulk TCR分析最容易犯的错误,是把某一张图当成全部结论。
比如只看克隆扩增,你可能会觉得疾病组更“寡克隆”;只看Chao1,你又会觉得疾病组多样性更高;只看MDS,你可能会觉得两组分不开。
在实际案例中,合理的做法是,我们把这些指标合在一起看:例如MS组在这个数据里更像是一个“多样性更高、部分样本存在扩增、V基因使用有偏移、组内异质性更强”的免疫组库状态。
这也是为什么我觉得OmicVerse里把这些bulk AIRR分析函数统一起来是有意义的。对用户来说,不需要在多个R包和Python包之间来回切换,就能把一套免疫组库图表跑完。
如果你做的是疫苗、感染、肿瘤免疫治疗或者自身免疫病队列,这套流程基本都可以直接改样本分组后复用。
19. 交流群
如果你也很好奇我们omicverse生态的最新进展,想第一时间体验到新功能,欢迎加入我们的交流群~

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
