python单细胞流程-降维
- 2026-06-29 21:15:00
为什么要进行降维?
在上一节中,我们已经从所有基因中筛选出了高变基因。高变基因选择本身已经完成了一次“特征层面”的降维:
全部基因 → 高变基因
但是,即使只保留 2000 个高变基因,每个细胞仍然是一个 2000 维左右的表达向量。这样的高维空间中包含真实的细胞类型差异,也包含技术噪音、基因间冗余和 dropout 带来的随机波动。如果直接在这样的高维矩阵上计算细胞之间的距离,结果往往不稳定,也不利于后续聚类和可视化。
降维(dimensionality reduction)的目的,是把每个细胞从高维基因表达空间投影到较低维的表示中,同时尽量保留最主要的生物学结构。简单理解就是:
高变基因表达矩阵 → 少量主成分或二维坐标
在单细胞 RNA-seq 的常规流程中,降维通常包含三层含义:
使用 PCA 将高变基因表达矩阵压缩到几十个主成分,用于后续邻居图构建和聚类。 使用 UMAP 或 t-SNE 将细胞进一步投影到二维空间,主要用于可视化。 检查低维空间是否被技术因素驱动,例如总 UMI 数、检测基因数、线粒体比例或批次信息。
需要注意的是,PCA、UMAP 和 t-SNE 的用途并不完全相同。PCA 得到的几十个 PC 通常用于后续分析;UMAP 和 t-SNE 更多用于观察数据结构和展示结果,不能简单把二维图上的距离、方向或 cluster 大小当成严格的定量结论。
降维的基本流程
在 Scanpy 中,一个常见的降维流程是:
HVG 数据 → scaling → PCA → 邻居图 → UMAP / t-SNE
其中,PCA 是核心步骤。UMAP 在 Scanpy 中并不是直接对原始表达矩阵运行,而是先基于 PCA 空间构建细胞邻居图,再把邻居图嵌入到二维空间中。因此,UMAP 的结果会受到前面几个选择的影响,包括高变基因数量、是否 scaling、保留多少个 PC、n_neighbors 以及 min_dist 等参数。
近年的单细胞最佳实践仍然推荐将 PCA 作为标准基线。对于普通 PBMC 这类数据,通常保留 30 到 50 个 PC 已经足够捕捉主要细胞类型差异。UMAP 和 t-SNE 仍然是最常用的二维可视化方法,但现在更强调:二维嵌入主要帮助我们看局部邻域和整体结构线索,而不是替代统计检验或生物学验证。
读取高变基因筛选后的数据
import os
import numpy as np
import scanpy as sc
adata = sc.read_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/04_pbmc_10k_hvg.h5ad")
adata

这里读入的是上一节保存的高变基因筛选结果。如果上一节已经执行:
adata = adata[:, adata.var["highly_variable"]].copy()
那么当前 adata.X 中通常只剩高变基因。可以先确认一下对象的维度:
adata.shape
如果保留了 highly_variable 标记,也可以查看当前矩阵中高变基因的数量:
adata.var["highly_variable"].value_counts()
如果这里所有基因都是 True,说明上一节保存前已经完成了 subset。这样后续 scaling 和 PCA 都只会在高变基因上进行。
Scaling
PCA 会寻找数据中方差最大的方向。如果不同基因的表达尺度差异很大,高表达基因可能会对 PCA 产生更强影响。为了让每个基因在 PCA 中处于相对可比的尺度,通常会在 PCA 前进行 scaling。
Scaling 的作用是对每个基因做标准化:
其中, 表示第 个细胞中第 个基因的表达值, 是基因 在所有细胞中的平均表达, 是该基因的标准差。经过 scaling 后,每个基因的均值接近 0,标准差接近 1。
在 Scanpy 中可以使用:
sc.pp.scale(adata, max_value=10)
这里的 max_value=10 表示将极端的 scaled value 截断到 10 以内,避免少数异常高表达值过度影响 PCA。
需要注意的是,sc.pp.scale 会修改 adata.X。因此,在前面的归一化章节中保存 adata.raw = adata.copy() 很重要。这样即使当前 adata.X 变成了 scaled expression,后续查看 marker gene 表达时,仍然可以从 adata.raw 中读取 Log(CP10k+1) 表达值。
如果数据集非常大,不建议在完整基因矩阵上直接 scaling,因为这会显著增加内存压力。通常应该先完成高变基因筛选,再只对高变基因矩阵进行 scaling 和 PCA。
PCA 降维
PCA(principal component analysis,主成分分析)是一种线性降维方法。它会把原来的高变基因表达矩阵转换成一组新的变量,称为 principal components,简称 PCs。
每一个 PC 都是多个基因表达值的线性组合。PC1 捕捉数据中方差最大的方向,PC2 捕捉剩余方差中第二大的方向,并且与 PC1 正交。后面的 PC 依次捕捉越来越少的方差信息。
简单理解就是:
2000 个高变基因 → 50 个 PC
运行 PCA:
sc.pp.pca(
adata,
n_comps=50,
svd_solver="arpack",
random_state=0
)
这里的 n_comps=50 表示先计算 50 个候选 PC,并不代表后续分析一定会全部使用这 50 个 PC。后面构建邻居图时,会通过 n_pcs 指定真正使用多少个 PC。例如本教程后面使用 n_pcs=30,意思是只使用前 30 个 PC 来计算细胞之间的距离。
这样做的好处是:先多计算一些 PC,方便后面画 elbow plot 和累计解释方差;等判断完哪些 PC 比较有信息量之后,再决定实际用于邻居图和聚类的 PC 数量。
执行完成后,PCA 结果会被保存到 AnnData 对象中:
adata.obsm["X_pca"]:每个细胞在 PCA 空间中的坐标。adata.varm["PCs"]:每个基因在不同 PC 上的 loading。adata.uns["pca"]["variance"]:每个 PC 解释的方差。adata.uns["pca"]["variance_ratio"]:每个 PC 解释的方差比例。
可以查看 PCA 坐标:
adata.obsm["X_pca"].shape
如果这里得到的是:
(细胞数, 50)
说明每个细胞已经从高变基因空间被表示成了 50 维 PCA 坐标。
查看 PCA 结果
可以先画出 PC1 和 PC2,并用一些 QC 指标上色:
sc.pl.pca(
adata,
color=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
ncols=3
)
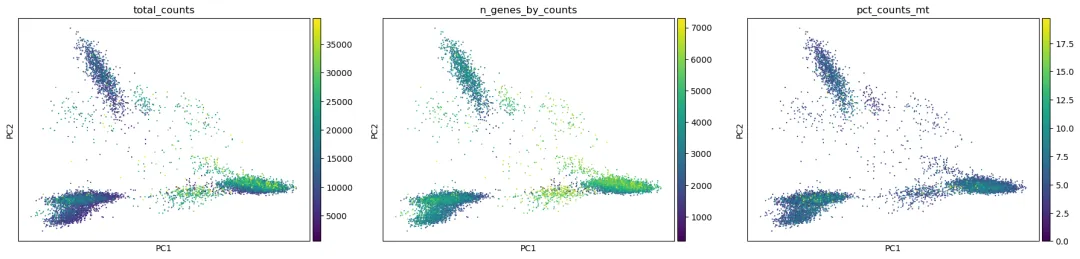
这一步的目的不是给细胞类型下结论,而是检查 PCA 是否明显被技术因素驱动。
如果 PC1 或 PC2 上的分离主要对应 total_counts高低,说明测序深度仍然可能影响较大。如果某一片细胞主要由高 pct_counts_mt细胞组成,说明低质量细胞可能没有完全过滤干净。如果有 sample或batch信息,也应该用这些列给 PCA 或 UMAP 上色,检查是否存在明显批次效应。
如果对象中存在样本或批次信息,可以继续查看(本数据没有):
obs_cols = [col for col in ["sample", "batch"] if col in adata.obs.columns]
if len(obs_cols) > 0:
sc.pl.pca(adata, color=obs_cols, ncols=2)
else:
print("当前 adata.obs 中没有 sample 或 batch 列,跳过批次信息检查。")
如果这里没有输出图,通常说明当前数据对象中没有 sample 或 batch 这类列。可以先查看所有细胞注释列:
adata.obs.columns
对于单一样本的 PBMC 教程数据,没有批次列是正常的。多样本数据通常需要在读入或合并样本时手动添加 sample、batch 或类似列。
选择保留多少个 PC
PCA 会计算很多个 PC,但后续构建邻居图时通常只使用前面一部分。保留太少的 PC 可能丢失真实生物学差异;保留太多的 PC 则可能把噪音也带入后续分析。
可以通过方差解释比例图观察每个 PC 的贡献:
sc.pl.pca_variance_ratio(adata, n_pcs=50, log=True)
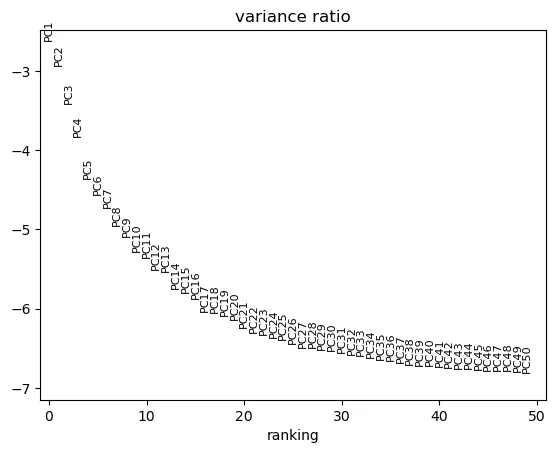
这张图通常也叫 elbow plot。判断时可以关注方差解释比例下降是否出现明显拐点。拐点之前的 PC 往往包含较多主要结构信息,拐点之后的 PC 通常贡献较小。
也可以计算前若干个 PC 的累计解释方差:
pca_var = adata.uns["pca"]["variance_ratio"]
for n_pcs in [10, 20, 30, 40, 50]:
print(n_pcs, np.sum(pca_var[:n_pcs]))
对于 PBMC 这类常规数据,实际分析中常从 n_pcs=30 开始。如果后续 UMAP 或聚类结果对 n_pcs 非常敏感,说明需要回头检查前面的 QC、HVG 选择、批次效应或样本组成。
如果看到类似下面的结果:
10 0.2319
20 0.2628
30 0.2793
40 0.2923
50 0.3038
可以理解为:相对于后面的 PC,前 10 个 PC 集中了更多方差信号;但这并不表示前 10 个 PC 已经解释了全部主要生物学差异。从 30 个 PC 增加到 50 个 PC,只额外增加了少量解释方差,说明 30 个 PC 可以作为一个合理起点,而不是最终答案。 单细胞数据中通常不需要像某些传统 PCA 场景那样,强行要求前几十个 PC 解释 80% 或 90% 的总方差。原因是单细胞表达矩阵非常稀疏、噪音较多,而且经过 scaling 后,每个高变基因的方差被拉到相近尺度,方差会分散到很多维度中。实际分析时更常见的做法是结合 elbow plot、后续 UMAP/聚类稳定性、QC 指标和 marker gene 结果,选择一个合适的 n_pcs。
本教程后续使用:
n_pcs = 30
构建邻居图
UMAP 需要先有一个细胞之间的邻居图。邻居图的基本思想是:在 PCA 空间中,对每个细胞找到最相似的一批细胞,然后记录这些局部邻域关系。
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=n_pcs,
random_state=0
)
这里的两个参数比较重要:
n_pcs=30:使用前 30 个 PC 计算细胞之间的距离。n_neighbors=15:每个细胞大约关注 15 个邻近细胞。较小的值更强调局部结构,较大的值会让结果更平滑、更偏向全局结构。
执行完成后,Scanpy 会在对象中保存邻居图信息:
adata.obsp["distances"]:细胞之间的邻居距离矩阵。adata.obsp["connectivities"]:细胞之间的连接强度矩阵。adata.uns["neighbors"]:邻居图相关参数。
可以查看:
adata.obsp["distances"].shape
adata.obsp["connectivities"].shape
这个邻居图不仅用于 UMAP,也会在下一章聚类时用于 Leiden 或 Louvain 聚类。因此,n_pcs 和 n_neighbors 不是单纯影响画图,也会影响后续分群结果。
UMAP 可视化
UMAP(uniform manifold approximation and projection)是一种非线性降维方法。它会尽量把高维邻居图中的局部关系保留到二维空间中,因此非常适合用来展示单细胞数据中的细胞群结构。
运行 UMAP:
sc.tl.umap(
adata,
min_dist=0.3,
spread=1.0,
random_state=0
)
画图查看:
sc.pl.umap(
adata,
color=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
ncols=3
)
这里先用 QC 指标上色,而不是直接解释细胞类型。原因是:如果 UMAP 上的主要分离由 total_counts、n_genes_by_counts 或 pct_counts_mt 驱动,那么后续聚类很可能也会受到技术因素影响。
如果想初步查看已知 marker gene 在 UMAP 上的分布,可以使用前面保存的 adata.raw:
marker_genes = ["MS4A1", "CD3D", "LYZ", "NKG7"]
if adata.raw isnotNone:
marker_genes = [gene for gene in marker_genes if gene in adata.raw.var_names]
sc.pl.umap(adata, color=marker_genes, use_raw=True, ncols=2)
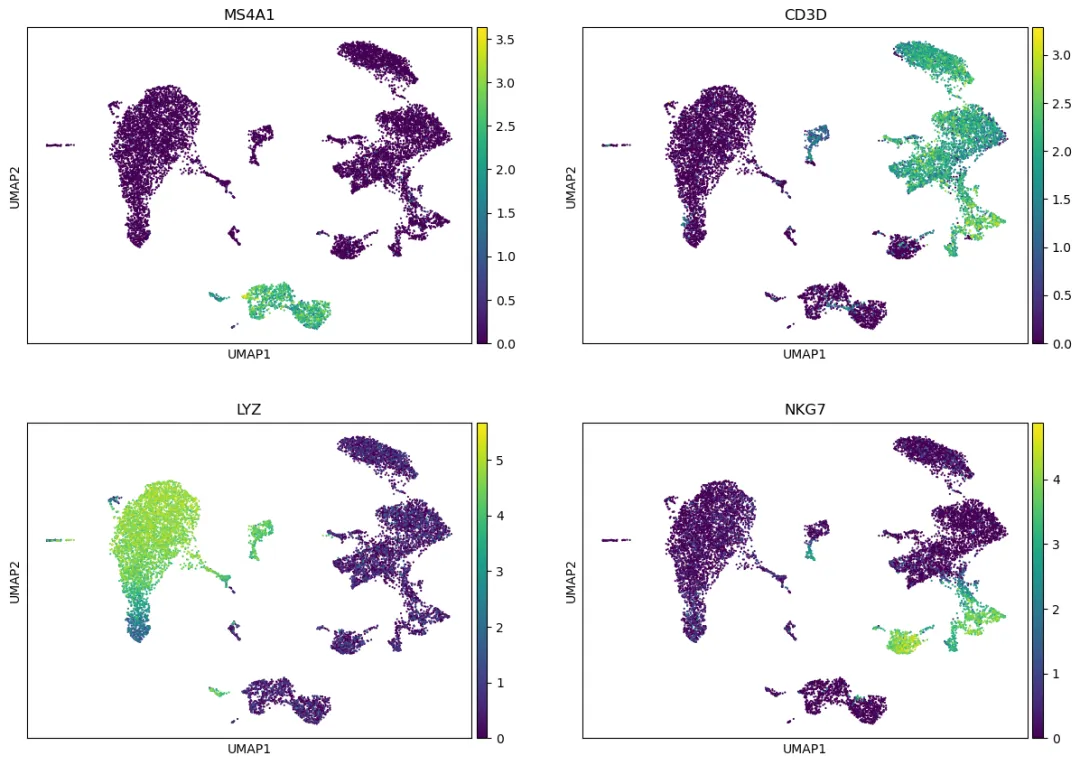
这里使用 use_raw=True,是为了显示 Log(CP10k+1) 后的表达值,而不是 scaling 后的 z-score。对于 marker gene 可视化,一般更推荐使用归一化后的表达值,而不是 scaled expression。
UMAP 结果会保存到:
adata.obsm["X_umap"]
可以查看:
adata.obsm["X_umap"].shape
如果结果是:
(细胞数, 2)
说明每个细胞已经有了二维 UMAP 坐标。
如何理解 UMAP 图?
UMAP 图上的每个点代表一个细胞。点与点之间越接近,通常表示它们在 PCA 邻居图中越相似。但在解释 UMAP 时需要特别谨慎。
UMAP 图可以帮助我们观察:
数据中是否存在明显的细胞群。 是否有连续变化的细胞状态。 某些 QC 指标、批次信息或 marker gene 是否集中在特定区域。 后续聚类结果是否和表达模式大致一致。
但是,UMAP 图不应该被这样解读:
不能因为两个 cluster 在 UMAP 上离得近,就直接认为它们在发育关系上更接近。 不能根据 UMAP 上某个 cluster 的面积判断细胞数量或细胞状态变异程度。 不能把 UMAP 的横轴或纵轴解释成具体生物学变量。 不能直接在 UMAP 二维坐标上做差异分析或统计检验。
近年的方法学研究反复提醒,t-SNE 和 UMAP 为了突出局部邻域结构,会不可避免地扭曲一部分全局距离。也就是说,UMAP 是非常有用的探索和展示工具,但它不是严格的几何地图。真正的生物学解释仍然需要结合 marker gene、样本信息、批次设计、差异表达分析和后续聚类结果。
t-SNE 可视化(可选)
t-SNE 也是单细胞分析中常见的二维可视化方法。它更强调局部邻域结构,常常能把不同细胞群分得很清楚,但计算速度通常比 UMAP 慢,对参数和随机种子也比较敏感。
在现在的 Scanpy 流程中,通常优先使用 UMAP。如果需要和已有文章或历史分析结果对比,也可以额外运行 t-SNE:
sc.tl.tsne(
adata,
n_pcs=n_pcs,
use_rep="X_pca",
perplexity=30,
random_state=0
)
画图:
sc.pl.tsne(
adata,
color=["total_counts", "n_genes_by_counts", "pct_counts_mt"],
ncols=3
)
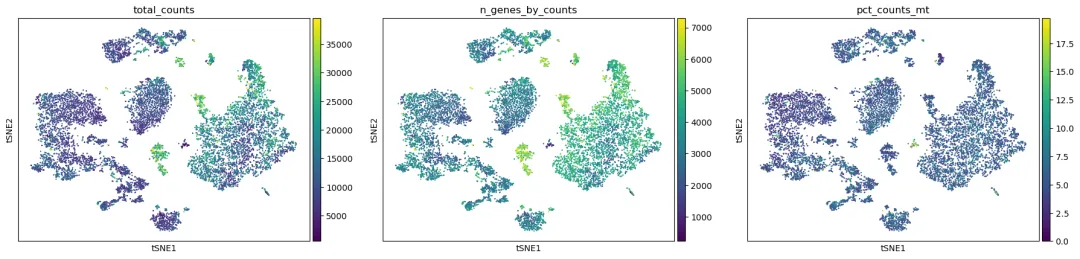
t-SNE 的解释原则和 UMAP 类似:主要看局部结构和细胞群分布,不要过度解释坐标轴、cluster 间距离或岛的大小。
检查降维结果是否合理
完成 PCA、邻居图和 UMAP 后,建议至少检查以下几类信息:
QC 指标
qc_cols = [col for col in ["total_counts", "n_genes_by_counts", "pct_counts_mt"] if col in adata.obs.columns]
sc.pl.umap(adata, color=qc_cols, ncols=3)
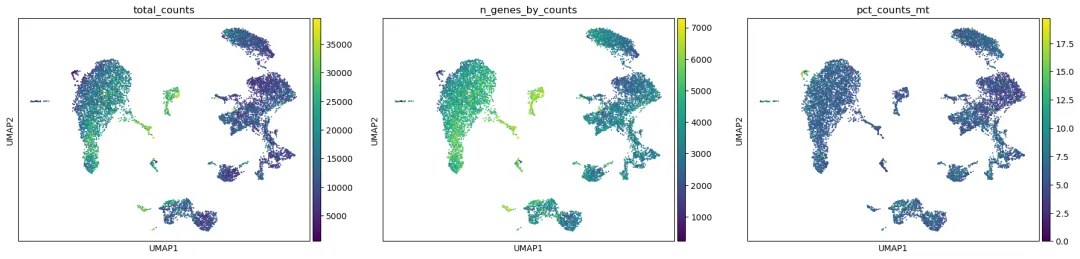
如果某些区域明显由低质量细胞、高线粒体比例细胞或极端 UMI 细胞组成,需要考虑回到质控步骤重新过滤。
样本或批次信息
batch_cols = [col for col in ["sample", "batch"] if col in adata.obs.columns]
if len(batch_cols) > 0:
sc.pl.umap(adata, color=batch_cols, ncols=2)
else:
print("当前 adata.obs 中没有 sample 或 batch 列,跳过批次信息检查。")
如果 UMAP 上主要按照样本或批次分开,而不是按照细胞类型分开,说明后续可能需要进行批次校正或数据整合。批次效应不应该简单通过调 UMAP 参数来“画得更好看”,而应该在合适的整合步骤中处理。
已知 marker gene
marker_genes = ["MS4A1", "CD3D", "LYZ", "NKG7", "PPBP"]
if adata.raw isnotNone:
marker_genes = [gene for gene in marker_genes if gene in adata.raw.var_names]
sc.pl.umap(adata, color=marker_genes, use_raw=True, ncols=3)
对于 PBMC 数据,常见 marker 可以帮助我们初步判断 UMAP 上的结构是否符合预期。例如:
MS4A1:B cells。CD3D:T cells。LYZ:monocytes。NKG7:NK cells 或 cytotoxic T cells。PPBP:platelets。
这一步只是初步检查,不等同于正式细胞类型注释。正式注释通常需要结合聚类、marker gene 差异表达和已有生物学知识。
降维和聚类参数怎么理解?
从高变基因到 UMAP 和聚类,中间有几个参数经常一起出现。它们不是彼此独立的,而是一条链:
n_comps → n_pcs → n_neighbors → UMAP min_dist → Leiden resolution
这里需要先区分两类参数。
第一类参数会影响邻居图,因此会同时影响 UMAP 和后续聚类:
n_pcs:使用多少个 PC 来计算细胞之间的距离。n_neighbors:每个细胞参考多少个邻近细胞来构建邻居图。
第二类参数主要影响可视化或聚类标签本身:
min_dist:主要影响 UMAP 图上点的紧密程度,不直接改变 Leiden 聚类结果。resolution:直接影响 Leiden 聚类的粗细程度,不改变已经计算好的 UMAP 坐标。
n_comps 和 n_pcs
n_comps 是 PCA 阶段计算多少个 PC:
sc.pp.pca(adata, n_comps=50)
这里的 50 是“先算出来备用”。真正进入邻居图的是 n_pcs:
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=30)
因此,n_comps=50 和 n_pcs=30 并不矛盾。前者表示计算 50 个候选主成分,后者表示实际使用前 30 个主成分。
n_pcs 太少时,可能只保留最强的粗粒度差异,导致一些较细的细胞亚群混在一起。n_pcs 太多时,可能把后面较弱的噪音 PC 也带入邻居图,导致 UMAP 和聚类出现不稳定的小碎群。
常见做法是先看 elbow plot,再从 20、30、40 这几个值中选择。对于 PBMC 10k 这种常规数据,n_pcs=30 是一个合理起点。
n_neighbors
n_neighbors 控制构建邻居图时,每个细胞关注多少个邻近细胞:
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=30
)
这个参数决定的是“局部结构”和“整体结构”之间的平衡。
较小的 n_neighbors,例如 10 或 15,会更强调局部结构,UMAP 上的细胞群可能分得更开,后续聚类也可能更细。较大的 n_neighbors,例如 30 或 50,会更强调整体连续结构,UMAP 更平滑,后续聚类可能更粗。
因此,n_neighbors 不是单纯影响画图,它会改变邻居图,也会影响 Leiden 聚类。对于 PBMC 这类细胞类型相对清楚的数据,n_neighbors=15 通常可以作为起点;如果数据更大、更连续,或者希望看到更平滑的整体结构,可以尝试 30。
UMAP 的 min_dist
min_dist 控制 UMAP 二维图上相邻点可以靠得多近:
sc.tl.umap(
adata,
min_dist=0.3,
spread=1.0,
random_state=0
)
较小的 min_dist 会让局部点更紧密,cluster 看起来更像一个个分开的岛;较大的 min_dist 会让点分布更松散,更容易看到连续过渡。
需要注意的是,如果 Leiden 聚类是基于 sc.pp.neighbors 得到的邻居图运行的,那么 min_dist 不会改变聚类结果。它主要影响 UMAP 的视觉形态。不要为了让图看起来“分得更开”,就把 min_dist 调得很小,然后把视觉分离直接当成新的细胞类型。
Leiden 的 resolution
Leiden 聚类是在邻居图上寻找细胞社区。它通常放在 UMAP 之后讲,是因为我们经常把聚类标签画在 UMAP 上,但从计算逻辑上说,Leiden 使用的是邻居图,不是 UMAP 二维坐标。
基本代码如下:
sc.tl.leiden(
adata,
resolution=0.5,
key_added="leiden_r0.5",
random_state=0
)
resolution 控制聚类的颗粒度:
较低的 resolution,例如 0.2 或 0.5,会得到更少、更大的 cluster。较高的 resolution,例如 1.0 或 1.5,会得到更多、更细的 cluster。
这个参数不等于“真实细胞类型数量”。它只是图聚类算法的分辨率。一个真实细胞类型可能被高分辨率拆成多个状态或亚群;多个相近细胞类型也可能在低分辨率下合并到同一个 cluster。
实际分析中,通常会同时尝试多个分辨率:
for res in [0.2, 0.5, 0.8, 1.0, 1.5]:
key = f"leiden_r{res}"
sc.tl.leiden(
adata,
resolution=res,
key_added=key,
random_state=0
)
print(key, adata.obs[key].nunique())
然后把不同分辨率的结果画在同一张 UMAP 上比较:
sc.pl.umap(
adata,
color=["leiden_r0.2", "leiden_r0.5", "leiden_r1.0"],
ncols=3
)
判断分辨率是否合适时,不应该只看 cluster 数量,而应该结合 marker gene 和生物学解释:
如果一个 cluster 内部明显包含两套互斥 marker,可能说明分辨率太低,需要拆开。 如果多个小 cluster 的 marker 几乎完全一样,可能说明分辨率太高,需要合并解释。 如果一个小 cluster 主要由高线粒体比例、极端 UMI 或某个批次组成,要优先考虑技术因素,而不是急着解释成新细胞类型。
对于 PBMC 教程数据,可以先从 resolution=0.5 或 resolution=0.8 开始。下一章正式聚类时,可以把多个分辨率都保存到 adata.obs 中,再结合 marker gene 选择最终用于注释的聚类结果。
推荐的调参顺序
实际分析中,建议按下面的顺序调整参数:
先固定高变基因选择和 scaling,运行 PCA。 根据 elbow plot 和累计解释方差选择 n_pcs,例如 20、30 或 40。在选定 n_pcs后构建邻居图,先使用n_neighbors=15。运行 UMAP,使用默认或接近默认的 min_dist,例如 0.3 到 0.5。在同一个邻居图上尝试多个 Leiden resolution,例如 0.2、0.5、0.8、1.0、1.5。结合 marker gene、QC 指标和样本信息,决定是否需要调整 n_pcs、n_neighbors或聚类分辨率。
不要一开始同时大幅调整所有参数。否则很难判断结果变化到底来自 PC 数、邻居图、UMAP 展示,还是聚类分辨率。
大数据集的补充说明
对于几千到几万个细胞的数据,常规 Scanpy 流程通常已经足够。对于几十万到上百万细胞的数据,PCA、邻居图、UMAP 和聚类会成为主要计算瓶颈。
近年的工具开始更多地支持 GPU 加速。例如 rapids-singlecell 提供了和 Scanpy 类似的接口,可以在 GPU 上加速 PCA、邻居图、UMAP、聚类和部分预处理步骤。对于普通 PBMC 10k 教程数据没有必要引入 GPU;但如果后续分析 atlas 级别数据,可以考虑使用这类工具提高计算效率。
另外,对于多批次、多样本或 atlas 级别数据,除了标准 PCA 表示,也常见使用 scVI/scANVI 等深度生成模型学习低维 latent representation。它们更适合放在“数据整合”或“批次校正”章节中讨论。本节先掌握最基础、最透明、最容易排查问题的 PCA 和 UMAP 流程。
保存
保存完成降维后的对象:
adata.write_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/05_pbmc_10k_dimensionality_reduction.h5ad")
检查文件是否保存成功:
os.path.exists("/home/data/t090639/project/pbmc_10k/data/processed/05_pbmc_10k_dimensionality_reduction.h5ad")
保存后的对象中应至少包含:
adata.obsm["X_pca"]:PCA 坐标。adata.obsm["X_umap"]:UMAP 坐标。adata.obsp["distances"]:邻居距离矩阵。adata.obsp["connectivities"]:邻居连接矩阵。adata.uns["pca"]:PCA 方差信息。adata.uns["neighbors"]:邻居图参数。adata.uns["umap"]:UMAP 参数。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- Linux进程控制保姆级介绍:理论与实战
- CentOS 7 升级到 Rocky Linux 9 新手完全指南(一):升级前准备与环境检查
- Linux内核漏洞允许本地攻击者获得root权限
- 打开网页,3秒跑Python
- Python/财务自动化之合并报表债权债务抵消
- Linux Direct I/O 实现机制:绕过 Page Cache 的数据通路解析
- Linux下C语言写文件落盘:fflush/fsync正确用法以及经典坑点避坑指南
- Python上下文管理器与with语句:资源管理的优雅之道
- Linux 最新资讯 20260609——Asterinas 0.18发布:Rust编写、内存安全的替代OS;Canonical 试验 Ubuntu 26.10 的 x86-64-v3 包构建
- 别让代码“算”计了你:Python运算符的规矩与魔法
