python单细胞流程-注释
- 2026-07-01 04:45:40
为什么要进行细胞类型注释?
在上一节中,我们已经完成了 Leiden 聚类。此时每个细胞都有了一个 cluster 标签,例如 leiden_res0_5。但是,cluster 只是算法在邻居图上找到的细胞群,并不自动等同于真实细胞类型。
细胞类型注释(cell type annotation)的目的,是把这些表达模式相近的细胞群解释成有生物学意义的身份,例如:
cluster 0 → CD4 T cells
cluster 1 → CD14+ monocytes
cluster 2 → B cells
在单细胞 RNA-seq 分析中,注释是从“数学分群”走向“生物学解释”的关键步骤。没有注释,后续的差异分析、细胞比例比较、轨迹分析和细胞通讯分析都很难解释。
需要注意的是,细胞类型注释不是简单地把某个自动软件的输出复制到结果表中。当前更主流、也更稳妥的做法,是把三类证据合在一起判断:
已知 marker gene 在 cluster 中的表达模式。 每个 cluster 自己富集的差异表达基因。 自动注释或参考映射工具给出的候选标签。
当前主流的注释策略
在 Python 单细胞分析中,常见注释策略可以分成几类。
第一类是基于 marker gene 的人工注释。这是最基础、也最需要保留的一步。即使后面使用自动注释,也应该回到 marker gene 检查结果是否合理。
第二类是基于 cluster marker 的反向解释。也就是先用 rank_genes_groups 找出每个 cluster 相对于其他 cluster 上调的基因,再根据这些基因判断细胞类型。
第三类是自动注释。对于 PBMC 和免疫细胞数据,CellTypist 是目前 Python 中非常常用、上手成本较低的选择。它使用训练好的参考模型,根据全转录组表达模式预测细胞类型,并可以通过 majority_voting=True 在局部亚群中平滑标签。
第四类是参考映射或 label transfer。对于有高质量参考图谱的项目,例如大型组织图谱、多样本整合或跨批次查询数据,scANVI、scArches、Azimuth 这类方法更适合。它们不是单纯看几个 marker,而是把 query 数据映射到带标签的 reference 上,再转移标签。缺点是需要合适的参考数据、更多计算资源,通常也需要更严格的批次和样本设计。
本节按照下面这条主线进行:
Leiden cluster
→ marker gene 可视化
→ cluster marker 筛选
→ 手动注释
→ CellTypist 自动注释交叉验证
→ 最终保存 cell_type
这条流程可以直接接在上一节的 PBMC 聚类结果后运行,并且每一步都有可视化或表格结果用于复核。
读取聚类后的数据
import os
import pandas as pd
import scanpy as sc
from IPython.display import display
adata = sc.read_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/06_pbmc_10k_clustered.h5ad")
adata

这里读入的是上一节保存的聚类结果。这个对象应该已经包含:
adata.obs["leiden_res0_5"]:Leiden 聚类标签。adata.obsm["X_umap"]:UMAP 坐标。adata.raw:归一化后的完整基因表达矩阵。adata.layers["counts"]:原始 UMI count。
先检查这些内容是否存在:
print(adata.obs.columns)
print(adata.obsm.keys())
print(adata.layers.keys())
print(adata.raw isnotNone)
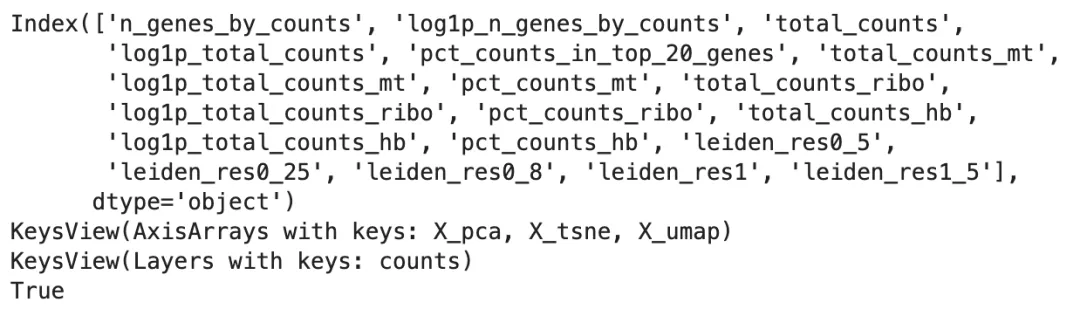
如果 adata.raw is not None 返回 True,说明前面保存的 Log(CP10k+1) 完整基因矩阵还在。后面展示 marker gene 和做 cluster marker 分析时,优先使用 adata.raw,因为当前 adata.X 可能已经只包含高变基因,甚至已经被 scaling。
选择一个主要用于注释的聚类结果:
cluster_key = "leiden_res0_5"
如果你在上一节发现 resolution=0.8 更适合当前数据,也可以改成:
cluster_key = "leiden_res0_8"
先看一下每个 cluster 的细胞数:
cluster_counts = adata.obs[cluster_key].value_counts().sort_index().to_frame("n_cells")
cluster_counts["pct_cells"] = cluster_counts["n_cells"] / adata.n_obs * 100
cluster_counts
非常小的 cluster 需要谨慎解释。它可能是真实稀有细胞类型,也可能是低质量细胞、doublet、批次效应或过高分辨率造成的小群。
用已知 marker gene 初步判断
对于 PBMC 数据,很多主要细胞类型有相对经典的 RNA marker。可以先准备一个 marker gene 字典。
pbmc_marker_genes = {
"CD4 T cells": ["CD3D", "CD3E", "IL7R", "CCR7", "LTB"],
"CD8 T cells": ["CD3D", "CD3E", "CD8A", "CD8B", "GZMK"],
"NK cells": ["NKG7", "GNLY", "KLRD1", "PRF1"],
"B cells": ["MS4A1", "CD79A", "CD79B", "CD74"],
"Plasma cells": ["MZB1", "JCHAIN", "XBP1", "SDC1"],
"CD14+ monocytes": ["LYZ", "S100A8", "S100A9", "FCN1", "CD14"],
"FCGR3A+ monocytes": ["FCGR3A", "MS4A7", "LST1", "IFITM3"],
"Dendritic cells": ["FCER1A", "CST3", "CLEC10A"],
"pDC": ["IL3RA", "GZMB", "TCF4", "IRF7"],
"Platelets": ["PPBP", "PF4", "GP9"],
"Erythroid cells": ["HBB", "HBA1", "HBA2", "ALAS2"],
}
这里需要注意两点。
marker gene 不是绝对的。不同组织、物种、平台、疾病状态和处理方式都会影响 marker 表达。 蛋白水平常用的 marker 不一定在 RNA 层面同样好用。例如某些 FACS marker 在 scRNA-seq 中可能 dropout 很严重。
为了避免基因不存在时报错,先只保留当前数据中能找到的 marker:
use_raw = adata.raw isnotNone
gene_names = adata.raw.var_names if use_raw else adata.var_names
marker_genes_in_data = {}
for cell_type, genes in pbmc_marker_genes.items():
found_genes = [gene for gene in genes if gene in gene_names]
if len(found_genes) > 0:
marker_genes_in_data[cell_type] = found_genes
marker_genes_in_data
如果很多经典 marker 都找不到,需要先检查基因 ID 是否是 gene symbol。如果矩阵中使用的是 Ensembl ID,很多自动注释模型和 marker gene 字典都不能直接匹配,需要先完成 ID 转换。
使用 dotplot 查看这些 marker 在不同 cluster 中的表达:
if len(marker_genes_in_data) > 0:
sc.pl.dotplot(
adata,
marker_genes_in_data,
groupby=cluster_key,
use_raw=use_raw,
standard_scale="var"
)
else:
print("当前数据中没有找到 marker gene,请检查基因名是否为 gene symbol。")
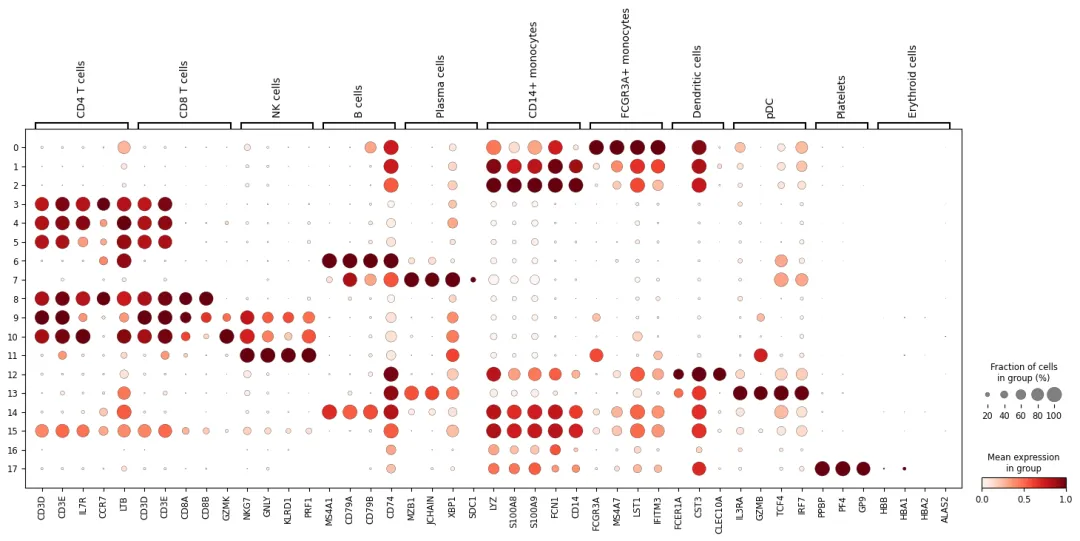
Dotplot 中有两个信息:
点的大小表示该 cluster 中表达这个基因的细胞比例。 颜色深浅表示该基因在不同 cluster 中的相对表达强度。
判断细胞类型时,不要只看单个基因。更稳妥的方式是看一组 marker 是否共同支持同一个身份。例如一个 cluster 同时高表达 CD3D、CD3E 和 IL7R,更支持 T cell;如果同时高表达 LYZ、S100A8 和 FCN1,更支持 CD14+ monocytes。
也可以把一些代表性 marker 画到 UMAP 上:
umap_marker_genes = [
"MS4A1", "CD3D", "CD4", "CD8A",
"LYZ", "FCGR3A", "NKG7", "PPBP"
]
umap_marker_genes = [gene for gene in umap_marker_genes if gene in gene_names]
if len(umap_marker_genes) > 0:
sc.pl.umap(
adata,
color=umap_marker_genes,
use_raw=use_raw,
ncols=4,
vmax="p99"
)
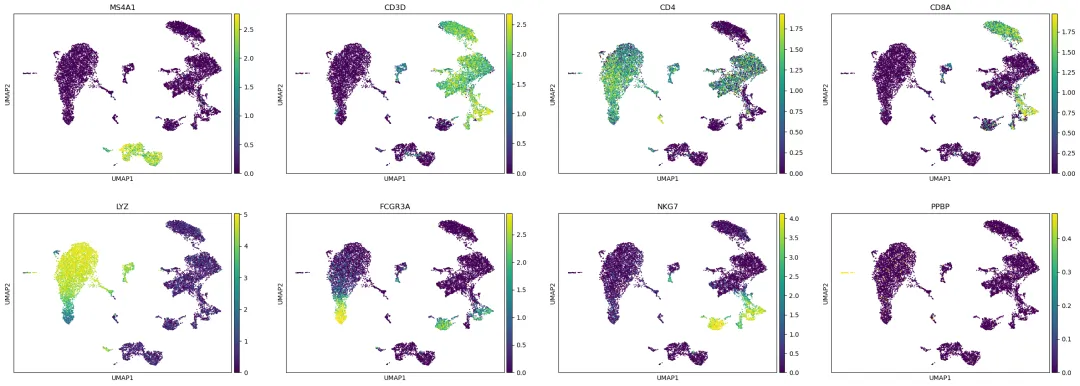
这里使用 vmax="p99" 是为了避免少数极端高表达细胞把颜色范围拉得太开。它不会改变数据,只影响图的显示。
给 marker gene 打分
除了直接看 dotplot,也可以给每组 marker 计算一个 module score。这个分数不能替代注释,但可以帮助快速比较每个 cluster 更像哪一类细胞。
score_cols = []
for cell_type, genes in marker_genes_in_data.items():
score_name = f"score_{cell_type}"
sc.tl.score_genes(
adata,
gene_list=genes,
score_name=score_name,
use_raw=use_raw
)
score_cols.append(score_name)
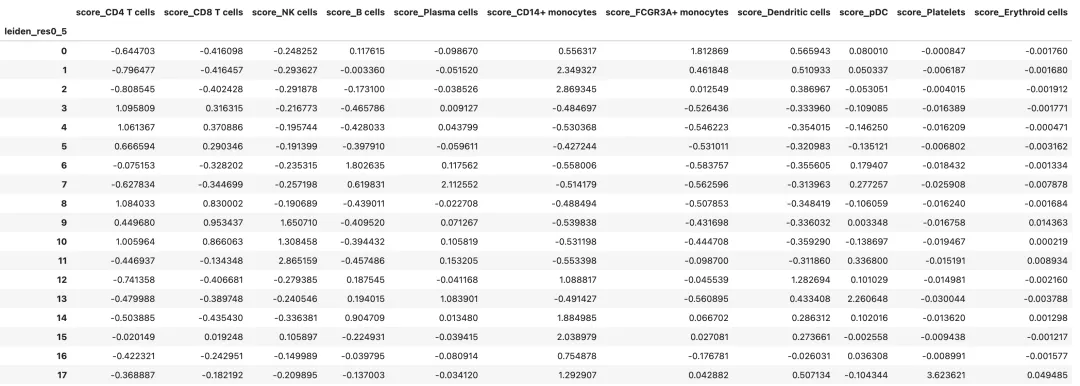
查看每个 cluster 的平均 marker score:
if len(score_cols) > 0:
marker_score_summary = adata.obs.groupby(cluster_key, observed=True)[score_cols].mean()
marker_score_summary
else:
print("没有可用的 marker score。")
在 notebook 中可以用颜色辅助查看:
if len(score_cols) > 0:
marker_score_summary.style.background_gradient(cmap="viridis", axis=0)

需要注意的是,module score 会受到 marker 数量、背景基因选择和数据归一化方式影响。它适合做辅助判断,不适合单独作为最终注释依据。
寻找每个 cluster 的 marker gene
上一节是从已知细胞类型出发,看这些 marker 出现在哪些 cluster 中。现在换一个方向:从每个 cluster 出发,找这个 cluster 相对于其他细胞上调的基因。
Scanpy 中可以使用 sc.tl.rank_genes_groups。这个函数要求输入是已经 log 转换的数据。因此,如果前面保留了 adata.raw,这里应使用 use_raw=True。
rank_key = f"rank_genes_{cluster_key}"
sc.tl.rank_genes_groups(
adata,
groupby=cluster_key,
method="wilcoxon",
use_raw=use_raw,
pts=True,
key_added=rank_key
)
这里几个参数的含义是:
groupby=cluster_key:按 Leiden cluster 寻找 marker。method="wilcoxon":使用 Wilcoxon rank-sum test,是单细胞 cluster marker 分析中常见的稳健选择。use_raw=use_raw:如果有adata.raw,使用完整的 Log(CP10k+1) 表达矩阵。pts=True:同时计算每个基因在组内和组外的表达细胞比例。key_added=rank_key:把结果保存到adata.uns[rank_key]中,避免覆盖其他分析。
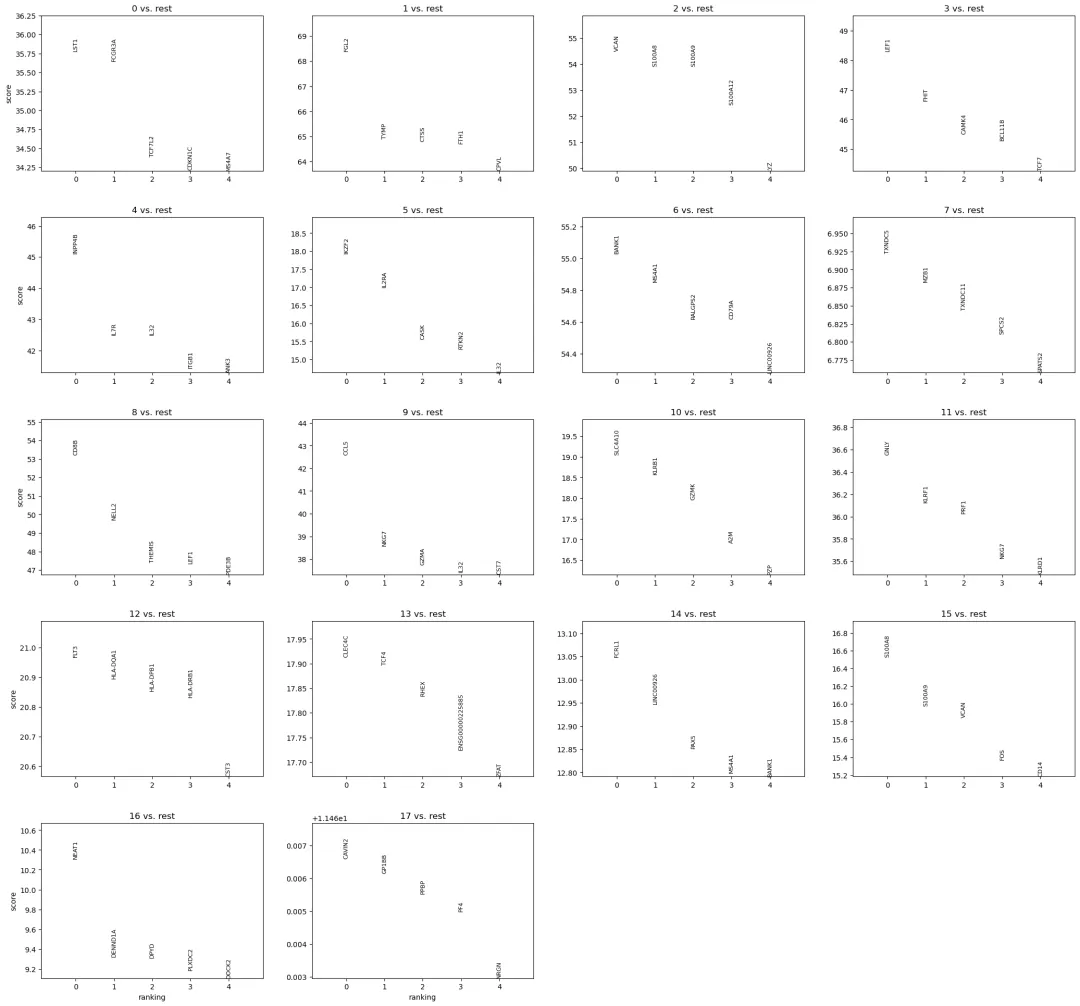
可以先画每个 cluster 排名前几的 marker:
sc.pl.rank_genes_groups(
adata,
key=rank_key,
n_genes=5,
sharey=False
)
也可以用 dotplot 形式展示:
sc.pl.rank_genes_groups_dotplot(
adata,
key=rank_key,
groupby=cluster_key,
n_genes=5,
standard_scale="var"
)
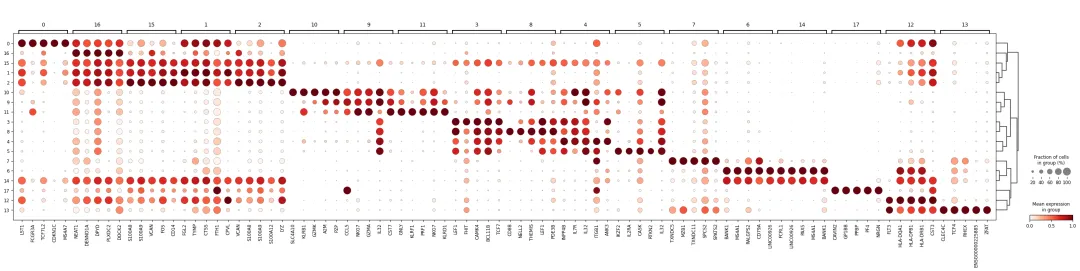
如果某个 cluster 排名前几的基因主要是线粒体基因、核糖体基因、血红蛋白基因或热休克基因,需要谨慎。它可能代表真实细胞类型,也可能提示低质量细胞、环境 RNA、应激状态或技术因素。
整理 cluster marker 表格
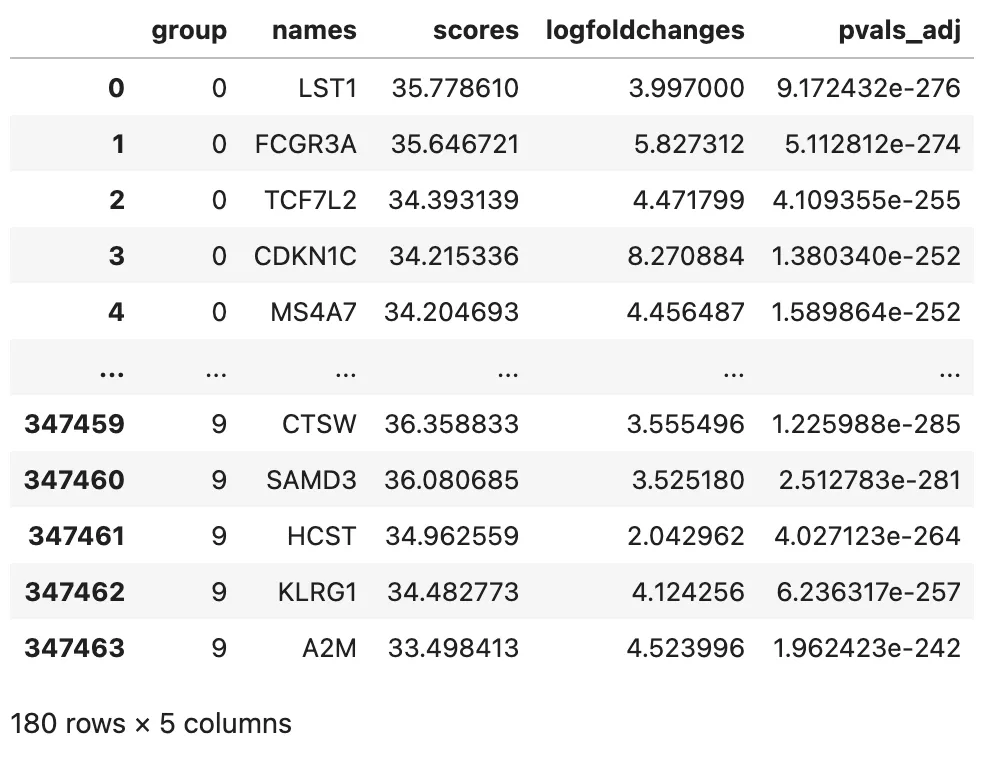
把 rank_genes_groups 的结果整理成表格,方便逐个 cluster 查看。
marker_df = sc.get.rank_genes_groups_df(
adata,
group=None,
key=rank_key
)
marker_df.head()
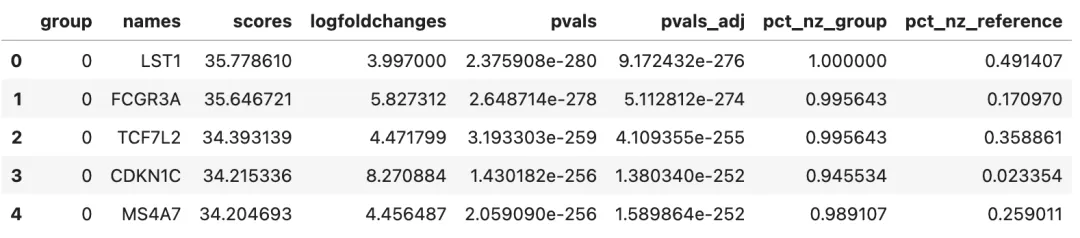
常见字段包括:
group:cluster 编号。names:基因名。scores:统计量。logfoldchanges:近似 log fold change。pvals:未校正 p 值。pvals_adj:多重检验校正后的 p 值。pct_nz_group:该 cluster 中表达该基因的细胞比例。pts_nz_reference:其他 cluster 中表达该基因的细胞比例。
可以按一些常用阈值过滤,得到更容易解释的 marker 表:
marker_df_filtered = marker_df.dropna(subset=["names"]).copy()
filter_mask = marker_df_filtered["pvals_adj"] < 0.05
if"logfoldchanges"in marker_df_filtered.columns:
filter_mask = filter_mask & (marker_df_filtered["logfoldchanges"] > 0.5)
marker_df_filtered = marker_df_filtered[filter_mask]
if {"pts", "pts_rest"}.issubset(marker_df_filtered.columns):
marker_df_filtered = marker_df_filtered[
(marker_df_filtered["pts"] > 0.25)
& (marker_df_filtered["pts"] > marker_df_filtered["pts_rest"])
]
marker_df_filtered.groupby("group").head(10)
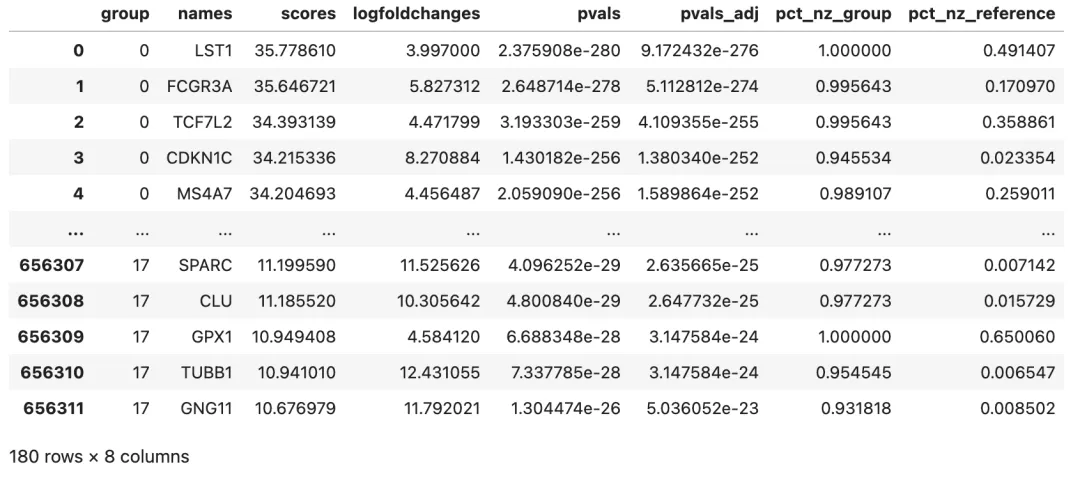
把每个 cluster 前 10 个 marker 单独列出来:
top_markers = (
marker_df_filtered
.sort_values(["group", "scores"], ascending=[True, False])
.groupby("group")
.head(10)
)
marker_display_cols = [
col for col in ["group", "names", "scores", "logfoldchanges", "pvals_adj"]
if col in top_markers.columns
]
top_markers[marker_display_cols]
如果想保存结果,方便后面查阅:
marker_out = "/home/data/t090639/project/pbmc_10k/data/processed/07_pbmc_10k_cluster_markers.csv"
top_markers.to_csv(marker_out, index=False)
os.path.exists(marker_out)
需要特别强调:这里的 cluster marker 是“细胞之间”的差异表达,细胞不是独立生物学重复。因此这个结果适合用于注释和探索,不适合直接作为严肃的条件间差异表达结论。真正比较不同样本或条件时,应该在后续章节使用 pseudobulk 或其他考虑样本重复的方法。
建立手动注释表
完成 marker gene 检查和 cluster marker 分析后,可以建立一个 cluster 到细胞类型的映射表。
为了让代码能直接运行,可以先把每个 cluster 默认命名为 Cluster x:
cluster_ids = sorted(
adata.obs[cluster_key].astype(str).unique(),
key=lambda x: (0, int(x)) if x.isdigit() else (1, x)
)
cluster_to_celltype = {cluster: f"Cluster {cluster}"for cluster in cluster_ids}
cluster_to_celltype
然后根据前面的 dotplot、UMAP marker 和 cluster marker 表,逐步修改这个字典。例如:
cluster_to_celltype.update({
"0": "FCGR3A+ monocytes",
"1": "CD14+ monocytes",
"2": "CD14+ monocytes",
"3": "CD4 T cells",
"4": "CD4 T cells",
"5": "CD4 T cells",
"6": "B cells",
"7": "Plasma cells",
"8": "CD8 T cells",
"9": "CD8 T cells",
"10": "CD8 T cells",
"11": "NK cells",
"12": "Dendritic cells",
"13": "pDC",
"14": "B/monocyte doublets",
"15": "CD14+ monocytes",
"16": "CD14+ monocytes",
"17": "Platelets",
})
把映射结果写入 adata.obs:
adata.obs["cell_type_manual"] = (
adata.obs[cluster_key]
.astype(str)
.map(cluster_to_celltype)
.astype("category")
)
adata.obs[[cluster_key, "cell_type_manual"]].drop_duplicates().sort_values(cluster_key)
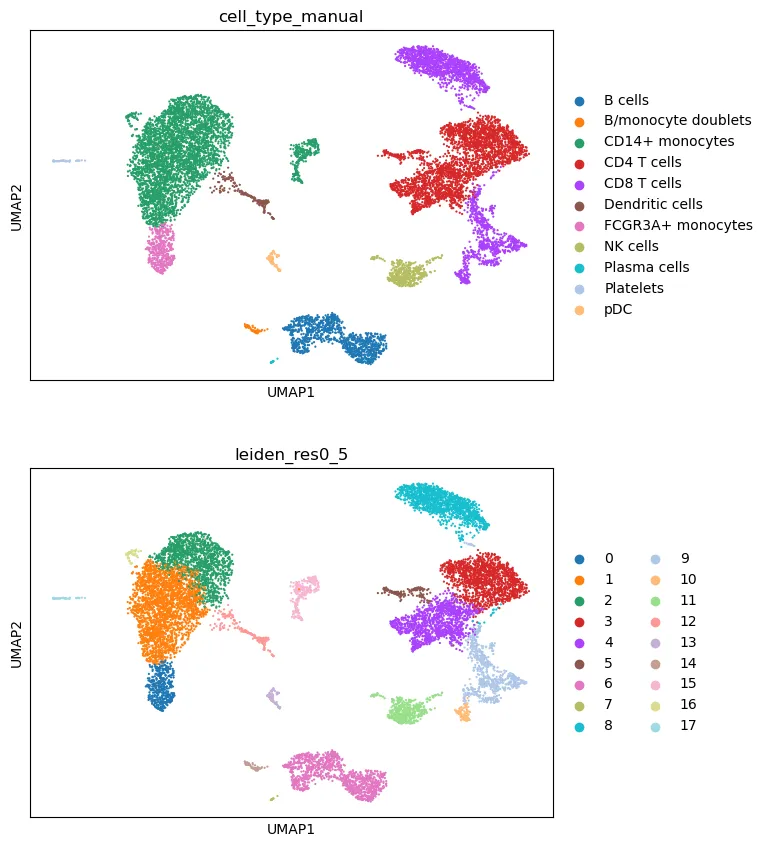
在 UMAP 上查看手动注释结果:
sc.pl.umap(
adata,
color=["cell_type_manual", cluster_key],
legend_loc="right margin",
ncols=1
)
再用 marker dotplot 检查注释后的细胞类型是否合理:
if len(marker_genes_in_data) > 0:
sc.pl.dotplot(
adata,
marker_genes_in_data,
groupby="cell_type_manual",
use_raw=use_raw,
standard_scale="var"
)
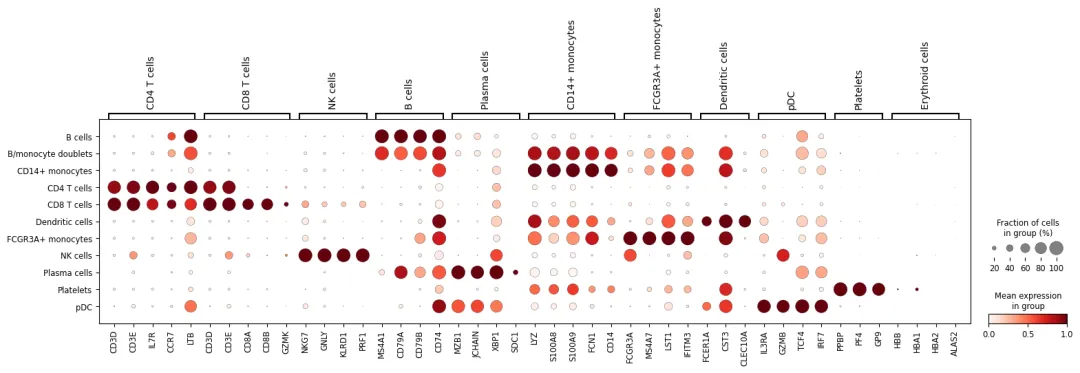
如果一个注释后的细胞类型内部同时高表达两套互斥 marker,例如同时高表达 T cell marker 和 monocyte marker,需要回头检查:
是否聚类分辨率过低,把不同细胞类型合并了。 是否存在 doublet。 是否 marker 选择不够具体。 是否该 cluster 其实是过渡状态或激活状态,而不是一个稳定细胞类型。
用 CellTypist 进行自动注释
对于 PBMC 这类免疫细胞数据,CellTypist 是非常常用的自动注释工具。它的优势是速度快、使用简单、内置免疫细胞参考模型。它的限制也很明确:预测结果依赖参考模型,如果 query 数据中有参考里没有的细胞类型或状态,结果可能会被强行分配到最相近的已有类型。
如果当前环境没有安装 CellTypist,可以先安装:
# pip install celltypist
导入包:
import celltypist
from celltypist import models
CellTypist 的输入应该是以 gene symbol 为基因名的表达矩阵,并且表达值应为每个细胞归一化到 10,000 counts 后的 log1p 表达。前面教程在归一化章节保存了:
adata.raw = adata.copy()
所以这里优先使用 adata.raw.to_adata(),避免误用已经 scaling 或只包含高变基因的 adata.X:
if adata.raw isnotNone:
adata_celltypist = adata.raw.to_adata()
elif"counts"in adata.layers:
adata_celltypist = adata.copy()
adata_celltypist.X = adata.layers["counts"].copy()
sc.pp.normalize_total(adata_celltypist, target_sum=1e4)
sc.pp.log1p(adata_celltypist)
else:
raise ValueError("CellTypist 需要 Log(CP10k+1) 全基因矩阵,或原始 counts layer。")
把已有的细胞元信息、UMAP 和邻居图复制过去,方便后续 majority voting 和可视化:
adata_celltypist.obs = adata.obs.copy()
for key in adata.obsm.keys():
adata_celltypist.obsm[key] = adata.obsm[key].copy()
for key in adata.obsp.keys():
adata_celltypist.obsp[key] = adata.obsp[key].copy()
if"neighbors"in adata.uns:
adata_celltypist.uns["neighbors"] = adata.uns["neighbors"].copy()
查看可用模型:
models.models_description()
对于 PBMC 数据,可以从免疫细胞模型开始。Immune_All_Low.pkl 是低层级、较细的免疫细胞标签模型,适合初步探索免疫细胞亚群。这里设置 force_update=True,让 CellTypist 先刷新模型索引,尽量使用当前可用的模型版本:
models.download_models(force_update=True, model="Immune_All_Low.pkl")
如果已经确认本地模型是最新的,也可以省略 force_update=True,减少重复联网:
# models.download_models(model="Immune_All_Low.pkl")
运行 CellTypist:
over_clustering_key = "leiden_res1"if"leiden_res1"in adata.obs.columns else cluster_key
celltypist_predictions = celltypist.annotate(
adata_celltypist,
model="Immune_All_Low.pkl",
majority_voting=True,
over_clustering=over_clustering_key
)
这里设置:
model="Immune_All_Low.pkl":使用免疫细胞细粒度模型。majority_voting=True:在局部亚群中进行多数投票,减少单细胞层面的噪音。over_clustering=over_clustering_key:优先使用已有较高分辨率 cluster 作为局部亚群。
把结果转回 AnnData,并复制到原始 adata.obs:
celltypist_adata = celltypist_predictions.to_adata()
adata.obs["celltypist_predicted_labels"] = (
celltypist_adata.obs.loc[adata.obs_names, "predicted_labels"]
.astype("category")
)
adata.obs["celltypist_majority_voting"] = (
celltypist_adata.obs.loc[adata.obs_names, "majority_voting"]
.astype("category")
)
adata.obs["celltypist_conf_score"] = celltypist_adata.obs.loc[
adata.obs_names,
"conf_score"
].astype(float)
可视化 CellTypist 结果:
sc.pl.umap(
adata,
color=[
"celltypist_majority_voting",
"celltypist_conf_score"
],
legend_loc="right margin",
ncols=1
)
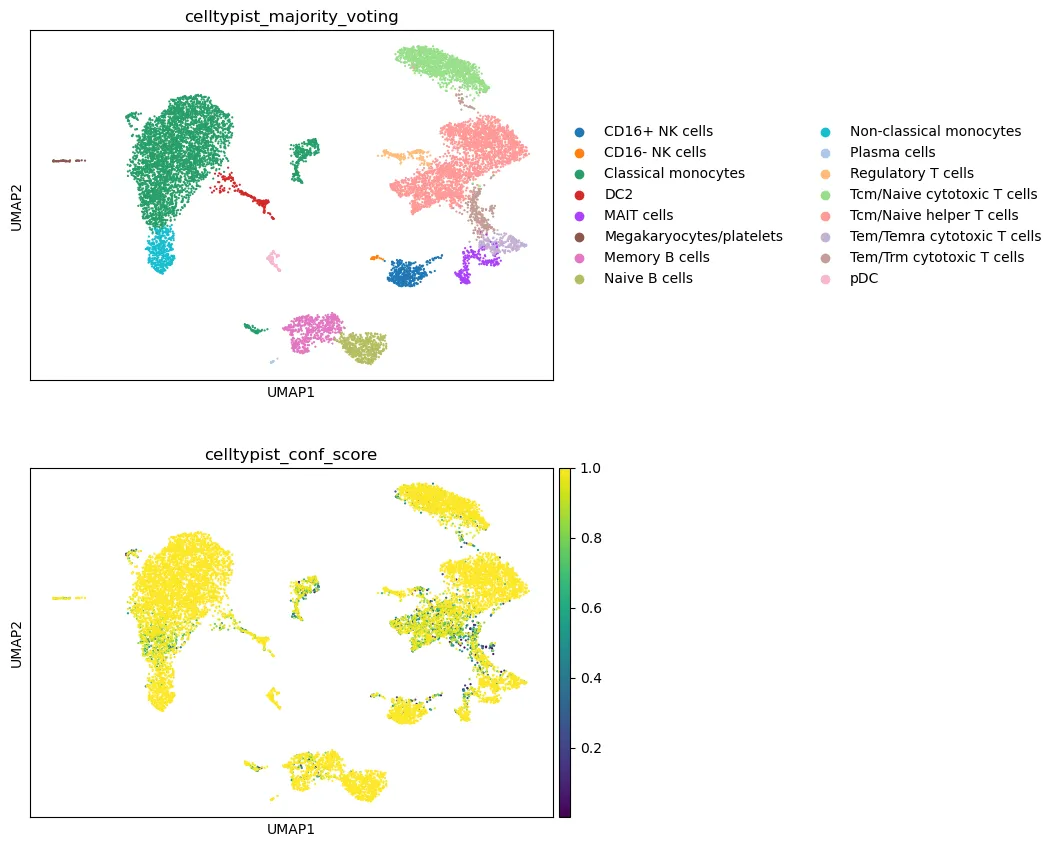
查看每个 cluster 中 CellTypist 标签的组成:
pd.crosstab(
adata.obs[cluster_key],
adata.obs["celltypist_majority_voting"],
normalize="index"
)
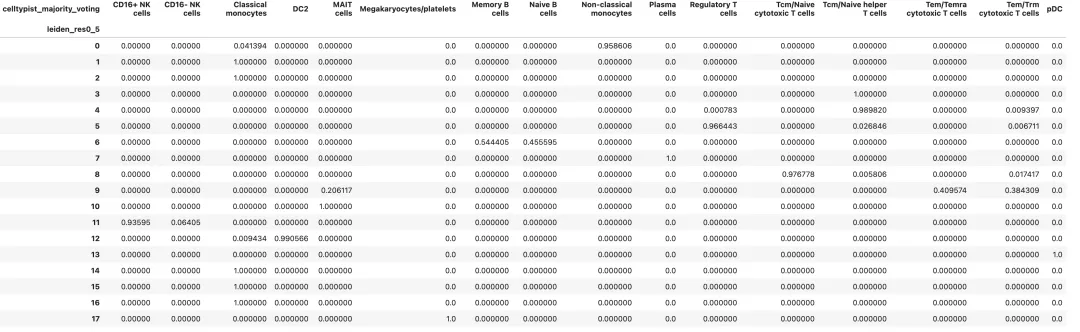
查看每个自动注释标签的置信度:
adata.obs.groupby(
"celltypist_majority_voting",
observed=True
)["celltypist_conf_score"].median().sort_values()
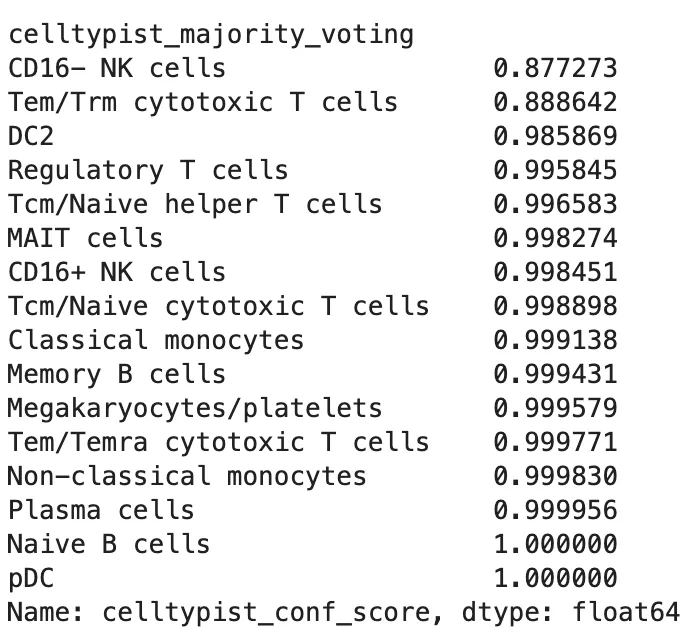
可以标记低置信度细胞:
adata.obs["celltypist_low_confidence"] = adata.obs["celltypist_conf_score"] < 0.5
adata.obs["celltypist_low_confidence"].value_counts()

这里的 0.5 不是通用真理,只是一个常用的初步筛查阈值。不同模型、组织和数据质量下,阈值都需要结合结果调整。低置信度不等于这个细胞一定错误,但它提示这个标签需要人工复核。
比较手动注释和自动注释
手动注释和自动注释通常不会完全一致。我们关心的不是让两个结果机械一致,而是找到需要复核的地方。
annotation_compare = pd.crosstab(
adata.obs["cell_type_manual"],
adata.obs["celltypist_majority_voting"],
normalize="index"
)
annotation_compare
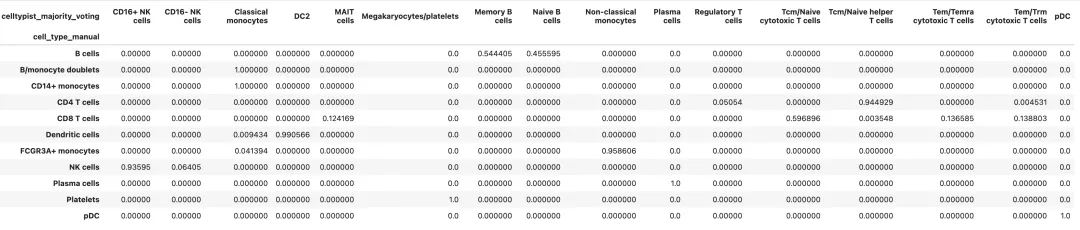
如果一个手动注释的细胞类型在 CellTypist 中被稳定预测为相近类型,说明二者互相支持。例如手动注释为 B cells,CellTypist 大多预测为 Naive B cells、Memory B cells 或 B cells,这通常比较合理。
如果一个手动注释的 cluster 被 CellTypist 分成很多不相关标签,或者 CellTypist 置信度整体很低,需要回到 marker gene 和 cluster marker 表重新检查。
还可以检查自动注释是否在表达空间中形成合理关系:
sc.tl.dendrogram(
adata,
groupby="celltypist_majority_voting",
use_raw=use_raw
)
sc.pl.dendrogram(
adata,
groupby="celltypist_majority_voting"
)
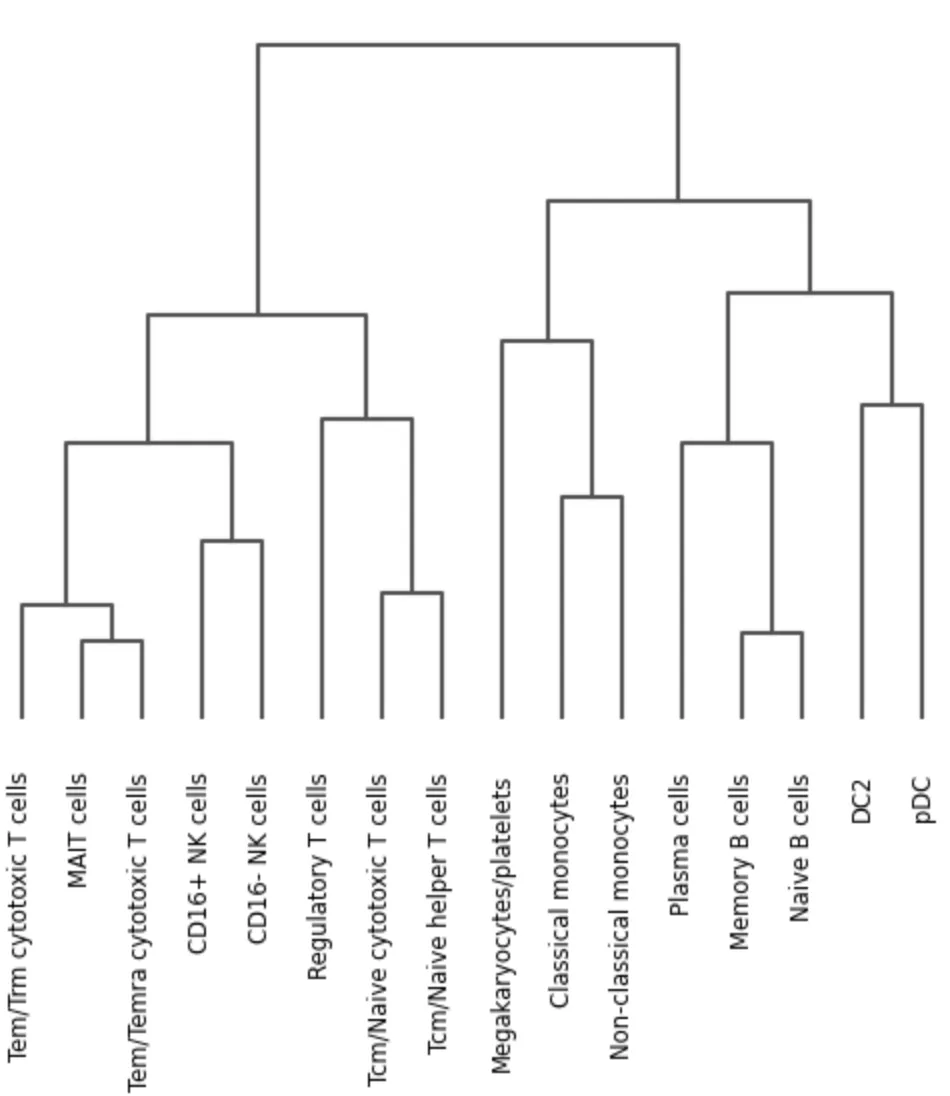
如果某个 B cell 子类在 dendrogram 中不和其他 B cell 靠近,或者某个 T cell 标签混在 monocyte 旁边,通常提示需要人工复核。
确定最终注释
最终注释建议以人工复核后的结果为主,自动注释作为辅助证据。可以把最终结果保存为统一列 cell_type:
adata.obs["cell_type"] = adata.obs["cell_type_manual"].astype(str)
adata.obs["cell_type"] = adata.obs["cell_type"].astype("category")
再保存一个注释置信度或复核状态列:
adata.obs["annotation_status"] = "manual_reviewed"
needs_review = (
adata.obs["cell_type"].str.startswith("Cluster")
| adata.obs["cell_type"].str.contains("doublet|ambiguous|unknown", case=False, regex=True)
| adata.obs.get("celltypist_low_confidence", False)
)
adata.obs.loc[needs_review, "annotation_status"] = "needs_review"
adata.obs["annotation_status"].value_counts()
这里的逻辑是:
已经根据 marker gene 明确命名的 cluster,标记为 manual_reviewed。仍然保留 Cluster x这种默认名称的 cluster,标记为needs_review。标记为 doublet、ambiguous 或 unknown 的 cluster,也标记为 needs_review。CellTypist 置信度低的细胞,也标记为 needs_review。
如果某些 cluster 的身份确实无法判断,不要强行命名。保留 Unknown、Ambiguous 或 Needs_review 往往比给出一个错误的细胞类型更好。
也可以建立粗粒度和细粒度两层注释。例如:
cell_type_to_major = {
"CD4 T cells": "T cells",
"CD8 T cells": "T cells",
"NK cells": "NK cells",
"B cells": "B cells",
"Plasma cells": "B cells",
"CD14+ monocytes": "Myeloid cells",
"FCGR3A+ monocytes": "Myeloid cells",
"Dendritic cells": "Myeloid cells",
"pDC": "Myeloid cells",
"Platelets": "Platelets",
"Erythroid cells": "Erythroid cells",
"B/monocyte doublets": "Needs review",
}
adata.obs["cell_type_major"] = (
adata.obs["cell_type"]
.astype(str)
.map(cell_type_to_major)
.fillna(adata.obs["cell_type"].astype(str))
.astype("category")
)
这种两层注释在后续分析中很有用。粗粒度标签适合做总体细胞组成比较,细粒度标签适合探索亚群。
注释后的质量检查
完成最终注释后,需要再次检查注释结果是否被技术因素驱动。
qc_plot_cols = [
col for col in ["total_counts", "n_genes_by_counts", "pct_counts_mt"]
if col in adata.obs.columns
]
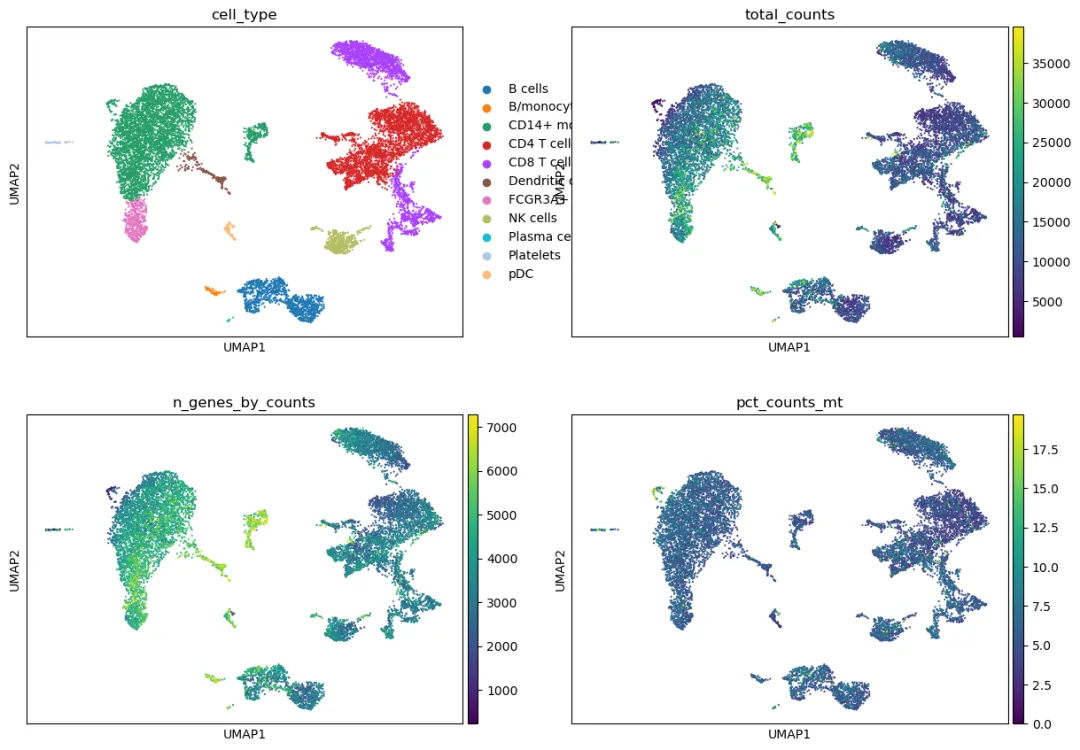
sc.pl.umap(
adata,
color=["cell_type"] + qc_plot_cols,
ncols=2
)
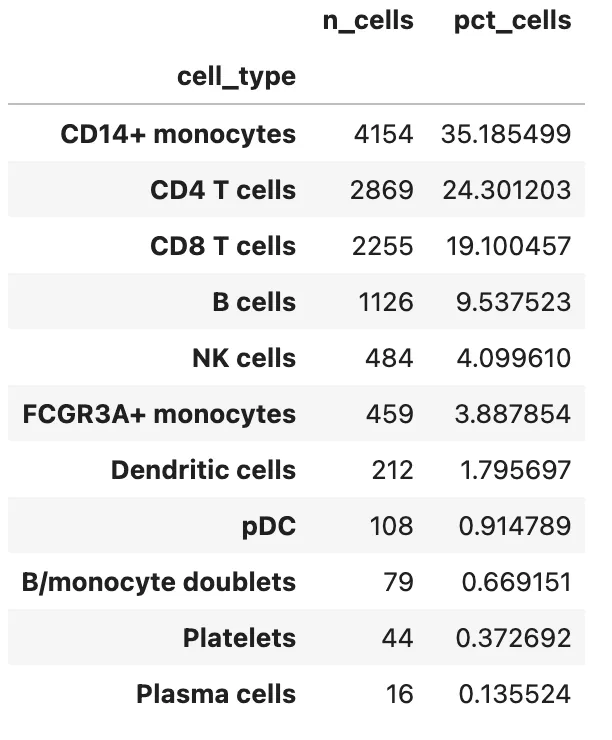
按最终细胞类型统计细胞数:
cell_type_counts = adata.obs["cell_type"].value_counts().to_frame("n_cells")
cell_type_counts["pct_cells"] = cell_type_counts["n_cells"] / adata.n_obs * 100
cell_type_counts
按最终细胞类型查看 QC 指标:
qc_cols = [
col for col in ["total_counts", "n_genes_by_counts", "pct_counts_mt"]
if col in adata.obs.columns
]
if len(qc_cols) > 0:
qc_summary = adata.obs.groupby("cell_type", observed=True)[qc_cols].median()
display(qc_summary)
else:
print("当前对象中没有找到常见 QC 指标列。")
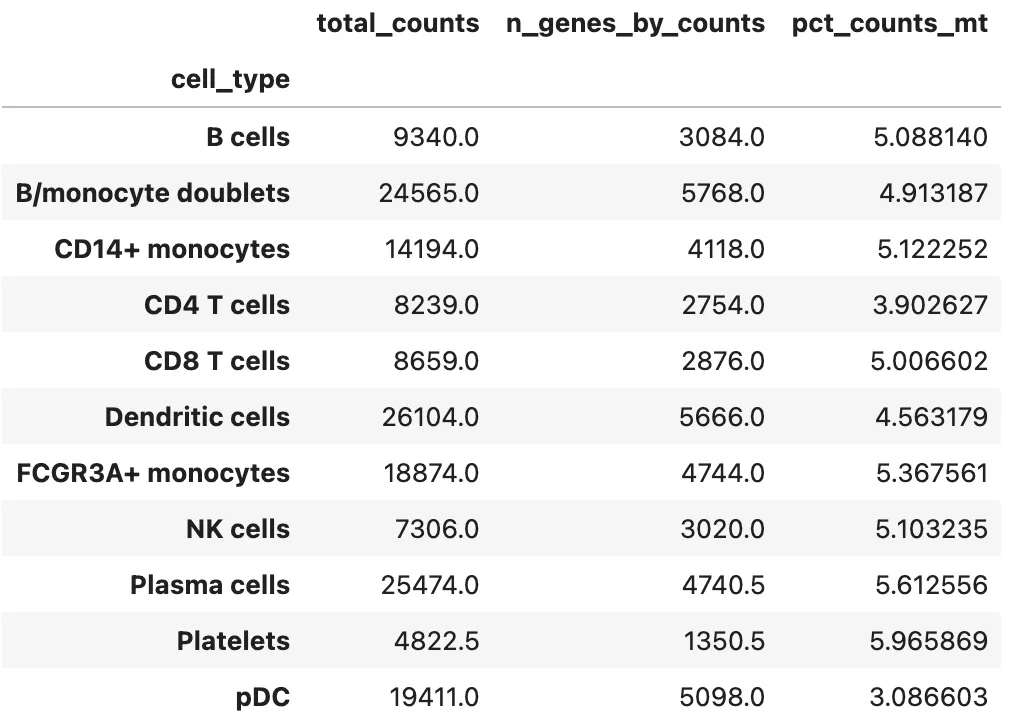
如果某个注释后的细胞类型几乎全部由高线粒体比例细胞组成,或者总 UMI 数极端异常,需要重新检查它是否是真实细胞类型。
如果对象中有样本或批次信息,也应该检查每个细胞类型是否被某个样本或批次完全主导:
sample_cols = [col for col in ["sample", "batch"] if col in adata.obs.columns]
if len(sample_cols) > 0:
for col in sample_cols:
print(col)
sample_table = pd.crosstab(
adata.obs["cell_type"],
adata.obs[col],
normalize="index"
)
display(sample_table)
else:
print("当前对象中没有找到 sample 或 batch 列,跳过样本/批次组成检查。")
对于多样本实验,如果某个细胞类型只出现在一个样本中,需要结合实验设计判断它是真实生物学差异,还是批次效应、样本质量差异或处理差异。
关于 scANVI 和参考映射
如果你有一个高质量、同物种、同组织或相近组织的参考图谱,并且参考中已经有可信的细胞类型标签,那么参考映射通常比单独使用 marker 字典更适合大规模项目。
在 Python 生态中,scvi-tools 的 scANVI 是当前主流选择之一。它是一个半监督模型,可以利用一部分已有细胞类型标签来推断未标注细胞的标签,也适合从已标注 atlas 向 query 数据转移标签。
这类方法适合以下场景:
多样本、多批次或跨平台数据。 需要把新数据映射到已有 atlas。 参考数据和 query 数据的组织、物种、平台足够接近。 有 GPU 或充足计算资源。
但它不适合在没有参考数据时盲目使用。参考映射不能发现参考中完全不存在的新细胞类型;如果 query 中有新的状态或疾病特异状态,模型可能会把它们分配到最相近的已有类型。因此,即使使用 scANVI、scArches 或 Azimuth,最终仍然需要回到 marker gene、cluster marker 和生物学背景进行验证。
对于当前这个 PBMC 入门教程,CellTypist 已经足够作为自动注释交叉验证。参考映射可以作为后续“多样本整合与参考图谱映射”章节再展开。
保存
保存完成注释后的对象:
adata.write_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/07_pbmc_10k_annotated.h5ad")
检查文件是否保存成功:
os.path.exists("/home/data/t090639/project/pbmc_10k/data/processed/07_pbmc_10k_annotated.h5ad")
保存后的对象中应至少包含:
adata.obs["cell_type"]:最终细胞类型注释。adata.obs["cell_type_manual"]:基于 marker 和 cluster 的人工注释。adata.obs["cell_type_major"]:可选的粗粒度细胞类型。adata.obs["annotation_status"]:注释复核状态。adata.obs["celltypist_majority_voting"]:CellTypist 自动注释结果。adata.obs["celltypist_conf_score"]:CellTypist 置信度。adata.uns[rank_key]:cluster marker 分析结果。
后续章节可以基于这里保存的 cell_type 继续做细胞类型比例分析、差异表达分析、轨迹推断或细胞通讯分析。

