anndataR | 一行代码读取h5ad为seurat对象
- 2026-07-09 04:22:33
anndataR | 一行代码读取h5ad为seurat对象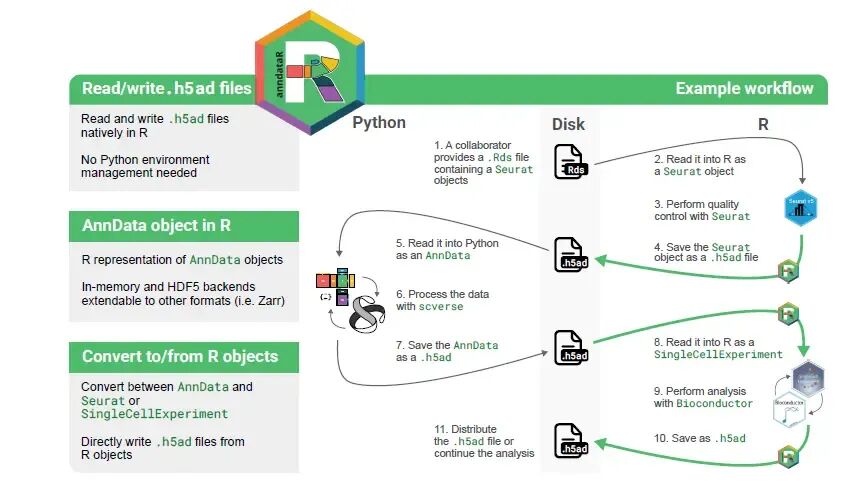
01 Seurat 02 SingleCellExperiment 03 Customizing 
04 h5ad 往期回顾 
squidpy | 空间转录组分析软件,scanpy团队出品 
从实现层详解scVelo的moments步骤让过程透明化 
scVelo分析RNA Velocity看似简单实则并不容易 
让bam满足velocyto的特别要求 
美化fgsea的GSEA分析结果 

软件的专业性最直接的体现方式就是使用起来是否简单高效。单细胞数据分析多了总会遇到一个问题,需要在R与python之间切换,而两种语言常用的保存格式却不一样,读写起来就显得不是很方便。今天分享一个R包可以直接将h5ad文件读取为seurat对象,过程相当简单,再也不用花费很多时间在格式转换上面了。
将h5ad文件读取为Seurat对象只一行命令即可搞定:
library(anndataR)h5ad_file <- system.file("extdata", "example.h5ad", package = "anndataR")obj <- read_h5ad(h5ad_file, as = "Seurat")objAn object of class Seurat100 features across 50 samples within 1 assayActive assay: RNA (100 features, 0 variable features) 5 layers present: counts, csc_counts, dense_X, dense_counts, X 2 dimensional reductions calculated: X_pca, X_umap在R里面单细胞数据很多软件的输入为SingleCellExperiment对象,将h5ad读取为该对象也是一行命令:
obj_sc <- read_h5ad(h5ad_file, as = "SingleCellExperiment")obj_scclass: SingleCellExperimentdim: 100 50metadata(18): Bool BoolNA ... rank_genes_groups umapassays(5): counts csc_counts dense_X dense_counts Xrownames(100): Gene000 Gene001 ... Gene098 Gene099rowData names(11): String n_cells_by_counts ... dispersions dispersions_normcolnames(50): Cell000 Cell001 ... Cell048 Cell049colData names(11): Float FloatNA ... log1p_total_counts leidenreducedDimNames(2): X_pca X_umapmainExpName: NULLaltExpNames(0):将h5ad文件读取为对象的时候,可以自定义这个过程来控制数据的生成,如生成自定义的Seurat对象:
adata <- read_h5ad(h5ad_file)adataInMemoryAnnData object with n_obs × n_vars = 50 × 100 obs: 'Float', 'FloatNA', 'Int', 'IntNA', 'Bool', 'BoolNA', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'leiden' var: 'String', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm' uns: 'Bool', 'BoolNA', 'Category', 'DataFrameEmpty', 'Int', 'IntNA', 'IntScalar', 'Sparse1D', 'String', 'String2D', 'StringScalar', 'hvg', 'leiden', 'log1p', 'neighbors', 'pca', 'rank_genes_groups', 'umap' obsm: 'X_pca', 'X_umap' varm: 'PCs' layers: 'counts', 'csc_counts', 'dense_X', 'dense_counts' obsp: 'connectivities', 'distances' varp: 'test_varp'obj <- adata$as_Seurat( layers_mapping = c("counts", "dense_counts"), object_metadata_mapping = c(metadata1 = "Int", metadata2 = "Float"), assay_metadata_mapping = FALSE, reduction_mapping = list(pca = c(key = "PC_", embeddings = "X_pca", loadings = "PCs"), umap = c(key = "UMAP_", embeddings = "X_umap")), graph_mapping = TRUE, misc_mapping = c(misc1 = "Bool", misc2 = "IntScalar"))obj每个mapping参数都可以设置为下面三种情况:
TRUE:默认slot里面的所有数据都会复制; FALSE:不复制slot里的数据; 命名字符向量:命令向量的元素名为对象里面slot名字,命令向量的元素为slot里面的数据。
将Seurat、SingleCellExperiment、AnnData保存为h5ad文件:
write_h5ad(obj, "sample.h5ad")在保存结果之前,也可以自定义AnnData的结果:
adata <- as_AnnData( obj, assay_name = "RNA", x_mapping = "counts", layers_mapping = c("dense_counts"), obs_mapping = c(RNA_count = "nCount_RNA", metadata1 = "metadata1"), var_mapping = FALSE, obsm_mapping = list(X_pca = "pca", X_umap = "umap"), obsp_mapping = TRUE, uns_mapping = c("misc1", "misc2"))--------- The End ---------
基因组
|
转录组
|
表观组
|
单细胞
寻找灵魂的工具人
长按扫码加关注
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

