R与Python用去偏LASSO模型、OW重叠加权、HDMA高维中介分析、SIS迭代筛选挖掘甲基化数据在童年虐待与PTSD关联介导机制与预测|附代码数据
全文链接:tecdat.cn/?p=44755原文出处:拓端数据部落公众号
关于分析师
在此对Yuan Rumeng对本文所作的贡献表示诚挚感谢,她在安徽理工大学完成了应用统计学专业的硕士学位,专注数据分析与人工智能领域。擅长R语言、Python、深度学习、数据挖掘与数据降维。Yuan Rumeng曾作为数据分析师,在多个涉及高维生物医学数据的项目中,利用统计建模与机器学习方法,深入挖掘疾病风险因子并构建预测模型,为临床研究的假设生成与证据转化提供了坚实的数据洞察支持。
项目文件目录截图:
在精准医疗时代,表观遗传数据已成为解析“环境-基因-疾病”复杂网络的核心钥匙(点击文末“阅读原文”获取完整智能体、代码、数据、文档)。
我们面临着前所未有的数据挑战:数十万个DNA甲基化位点与有限的临床样本并存,传统的“一因一果”分析框架已然失效。如何从这海量的噪声中,筛检出真正介导疾病发生的关键分子路径?
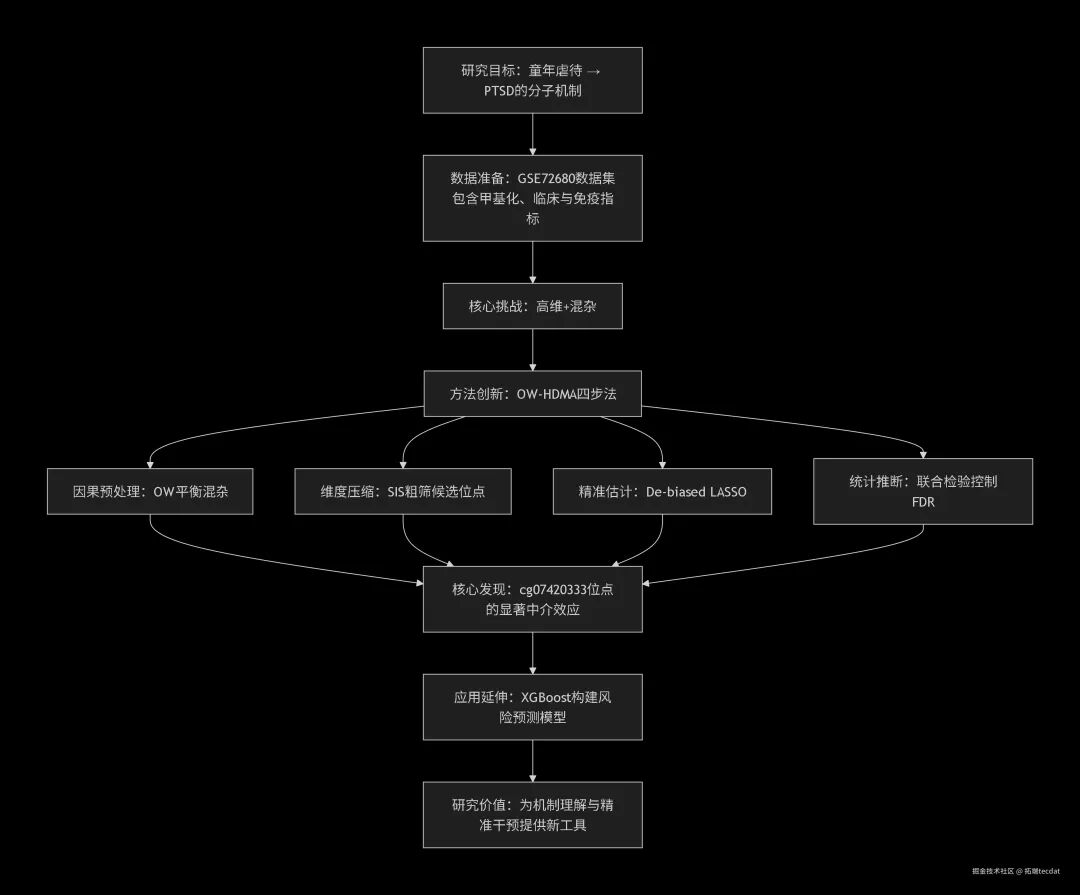
本文要讲述的,正是这样一个数据科学家如何运用统计智慧和计算工具,破解童年逆境如何“刻入”基因组并导致成年后心理创伤的故事。童年虐待是PTSD的已知风险因素,但其间精确的生物学桥梁一直是个黑箱。我们尝试用数据建立这座桥梁——不是简单关联,而是严谨的因果推断。
为此,我们设计了一套名为“OW-HDMA”的组合算法:用重叠加权(Overlap Weighting, OW) 平衡混杂,模拟随机试验的纯净环境;用高维中介分析(High-Dimensional Mediation Analysis, HDMA) 框架,结合迭代特征筛选(Sure Independence Screening, SIS) 与去偏LASSO估计,对三十余万个候选位点进行“大海捞针”式的精准捕捞。这套方法不仅解决了超高维数据的计算难题,更提升了在观察性研究中因果推断的稳健性。
本文内容改编自过往客户咨询项目的技术沉淀并且已通过实际业务校验,该项目完整代码与数据已分享至交流社群。阅读原文进群,可与800+行业人士交流成长;还提供人工答疑,拆解核心原理、代码逻辑与业务适配思路,帮大家既懂怎么做,也懂为什么这么做;遇代码运行问题,更能享24小时调试支持。
下方流程图直观地呈现了本次研究的“解题思路”:
研究背景:童年创伤的“生物烙印”
童年虐待是一个严峻的全球性公共卫生问题,其影响深远,是多种精神障碍的强力风险因素。令人深思的是,早年遭受的创伤,其阴影可以跨越数十年,显著增加个体成年后罹患创伤后应激障碍的风险。这背后隐藏着一个关键的生物学问题:早期的心理社会压力,是如何在分子层面留下持久印记,并改变个体终身的健康轨迹的?
近年来的研究将目光投向了表观遗传学,特别是DNA甲基化。DNA甲基化如同基因这本“生命之书”上的铅笔注释,它不改变文字(基因序列)本身,却能决定哪些段落(基因)被阅读(表达)。童年虐待这类强烈的环境压力,可能像一只无形的手,在某些关键基因上擦除或添加了甲基化“注释”,从而永久性地改变大脑应对压力的方式,埋下PTSD的易感种子。
然而,验证这一假说面临巨大方法学鸿沟。全基因组甲基化扫描产生数十万个数据点(CpG位点),样本量往往只有几百。这种“维度远超样本”的高维数据场景,使得传统统计方法要么力不从心,要么结果极不可靠。更复杂的是,童年虐待并非随机发生,受虐待群体与未受虐待群体在性别、年龄、社会经济地位等多方面存在系统性差异(即混杂偏倚)。若不妥善处理这些混杂因素,任何发现的“关联”都可能是误导。
因此,本研究的目标清晰而富有挑战:开发一套能同时处理“高维数据”和“观察性研究混杂偏倚”的分析框架,从分子层面实证童年虐待通过DNA甲基化影响PTSD的因果路径。
数据处理:从原始文件到分析变量
数据来源与清洗本研究数据源自一项公开的创伤研究队列(GEO:GSE72680),包含了童年虐待经历、PTSD症状评分(BDI和PSS)、年龄、性别、六类免疫细胞比例,以及约48万个CpG位点的全基因组甲基化数据。
原始数据格式较为杂乱,不同变量分散在各列。我们的第一步是进行数据清洗与整合。核心思路是遍历数据框的每一列,根据列名或内容特征(如包含“cd8”、“cd4”等关键词)提取对应的数值。
# 示例:清洗并提取CD8 T细胞数据# 初始化一个空列表,用于存放从每一列中找到的“cd8”相关值cd8结果列表 <- vector("list", ncol(原始数据框))for (第i列 in1:ncol(原始数据框)) { # 使用正则表达式匹配,不区分大小写查找包含“cd8”的单元格 匹配到的值 <- 原始数据框[[第i列]][grepl("cd8", 原始数据框[[第i列]], ignore.case = TRUE)]if (length(匹配到的值) > 0) { cd8结果列表[[第i列]] <- 匹配到的值 } else { cd8结果列表[[第i列]] <- NA # 如果没找到,用NA填充 }}# 将列表名设置为原始列名,方便后续追踪names(cd8结果列表) <- colnames(原始数据框)# ... 省略后续将列表转换为数据框、以及处理CD4、BDI等其他8个变量的类似代码 ...
代码解读:这段R代码通过循环和模式匹配(grepl),像用筛子一样从原始数据的每一列中“筛”出我们需要的特定变量值,是数据整理中常见的“宽变长”思路。
我们将提取出的CD8、CD4、童年虐待(Abuse)、自然杀伤细胞(NK cells)、B细胞(Bcells)、单核细胞(Monocytes)、粒细胞(Granulocytes)、贝克抑郁量表(BDI)、创伤后应激障碍症状量表(PSS)共9个关键变量的数据,通过添加标识列(Marker)后进行行合并,最终得到一个整洁的、便于分析的数据集。
变量定义清晰定义每个变量的角色是因果分析的基础:
- 暴露变量(X)
- 结果变量(Y) :是否患有PTSD。我们综合BDI和PSS量表评分,设定更严格的复合标准:
PSS≥14 且 BDI≥14 判定为有症状;PSS≤7 且 BDI≤7 判定为无症状。 - 中介变量(M) :全部约48万个CpG位点的甲基化M值(由原始的β值转换而来,统计特性更优)。
- 协变量(C) :年龄(Age)、性别(Sex)以及六种免疫细胞的比例,这些被视为潜在的混杂因素。
核心方法:OW-HDMA算法详解
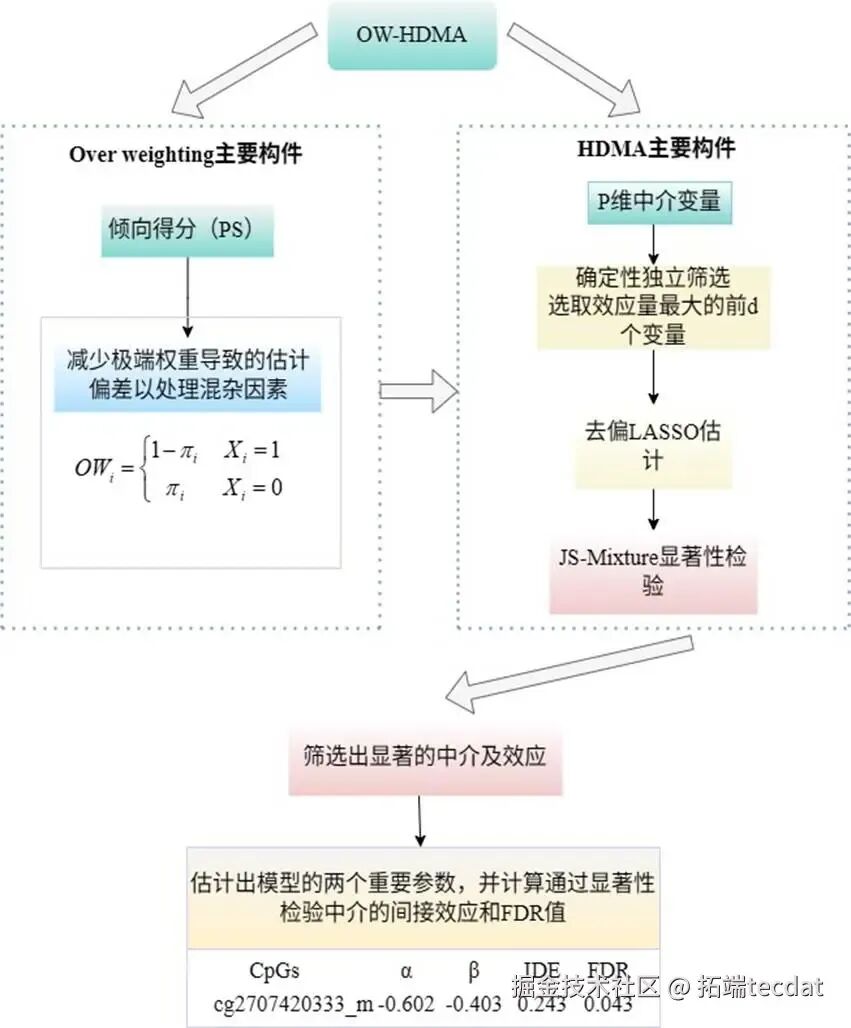
面对“超高维中介变量”和“非随机暴露”两大难题,我们创新性地将重叠加权整合进高维中介分析框架,形成四步走的稳健分析流程(OW-HDMA),其核心逻辑如图2所示。
第一步:用重叠加权(OW)平衡混杂,模拟随机试验在观察性研究中,直接比较“暴露组”(受虐)和“对照组”(未受虐)会因基线特征不平衡而产生偏倚。传统方法是逆概率加权,但容易因倾向评分极端而产生巨大方差。我们采用更稳定的重叠加权。它的思想很巧妙:更关注那些倾向评分(即基于协变量预测出的受虐待概率)在0.5附近的个体。这些人在两组中特征高度相似,如同随机试验中“被随机分到不同组”的个体,对他们的分析最能反映真实的暴露效应。
第二步:用迭代特征筛选(SIS)实现降维直接对48万个位点建模是不可能的。我们采用SIS方法,依据每个甲基化位点与结局(PTSD)的边际关联强度,快速筛选出前 d = [2n/log(n)] 个最相关的候选位点,将维度从数十万降至几十(本研究筛出78个),为后续精细分析铺路。
第三步:用去偏LASSO进行无偏系数估计在筛选出的候选位点中,位点间仍可能存在共线性(相关性)。我们使用去偏LASSO来拟合中介模型。它是LASSO的进阶版,能在进行变量选择、产生稀疏解的同时,对入选变量的系数进行纠偏,得到接近无偏的估计值及其标准误。
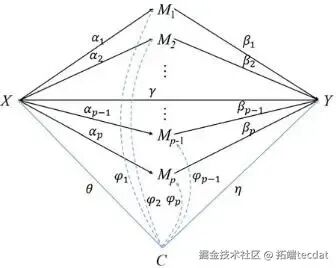
第四步:联合显著性检验与错误发现率控制对于每个候选位点,其中介效应等于“暴露→甲基化”的路径系数(α)与“甲基化→结局”的路径系数(β)的乘积。我们检验复合零假设 H₀: α=0 或 β=0(即至少一条路径不成立)。通过计算联合P值,并采用能精准控制错误发现率(FDR) 的混合零分布检验(JS-mixture) ,最终识别出那些通过严格统计检验的、真正发挥中介作用的甲基化位点。中介模型的因果路径如图3所示。
相关文章
Python梯度提升树、XGBoost、LASSO回归、决策树、SVM、随机森林预测中国A股上市公司数据研发操纵融合CEO特质与公司特征及SHAP可解释性研究
原文链接:tecdat.cn/?p=44265
结果分析:从免疫特征到分子中介
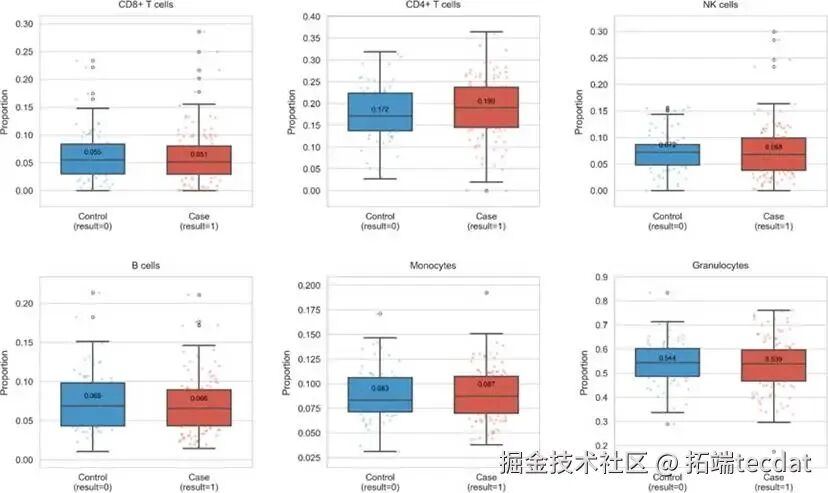
1. 研究人群基线特征我们首先比较了PTSD患者组与健康对照组的基线特征。如表1所示,两组在年龄、性别上无显著差异,但童年虐待经历的比例存在极显著差异(P < 0.001)。此外,部分免疫细胞的比例在组间也呈现出趋势性差异。
表1:病例组与对照组基线特征比较(部分)
图4更直观地展示了两组在六类免疫细胞比例分布上的差异,提示免疫系统状态可能与PTSD存在关联。
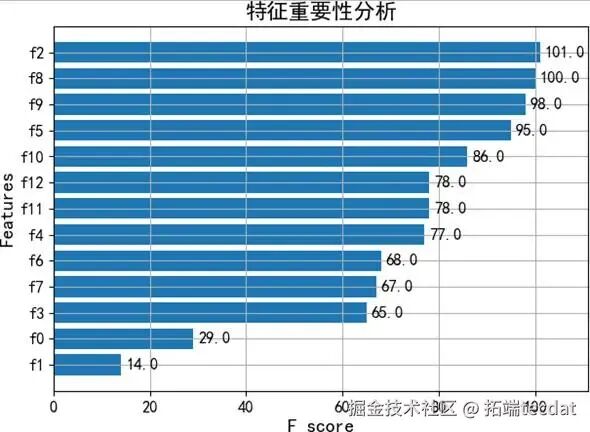
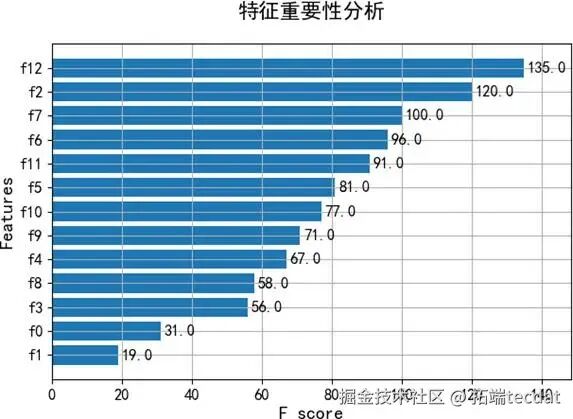
2. 基于XGBoost的抑郁与压力状态预测为探索利用现有变量预测心理状态的能力,我们构建了XGBoost模型,对根据BDI和PSS划分的抑郁/压力等级进行分类。模型表现良好,准确率达到88%。
# 定义XGBoost多分类模型的关键参数模型参数 = {'tree_method': 'gpu_hist', # 使用GPU加速,基于直方图算法构建树'objective': 'multi:softmax', # 指定为多分类任务'num_class': 4, # 目标类别数(针对BDI分为4类)'max_depth': 6, # 控制树的最大深度,防止过拟合'learning_rate': 0.1, # 学习率,控制每棵树的贡献权重'subsample': 0.8, # 每棵树训练时使用的样本比例'seed': 42 # 固定随机种子,确保结果可重现}# ... 省略数据标准化、转换为DMatrix格式、交叉验证训练及模型评估的详细代码 ...
模型的特征重要性分析(图5、图6)揭示了影响预测的关键因素,例如年龄和某些免疫细胞比例,这为理解影响心理状态的生物基础提供了线索。
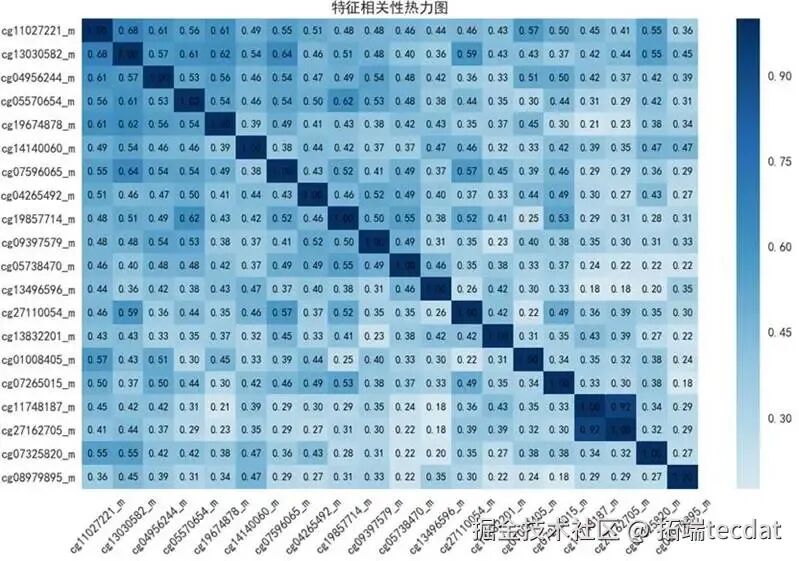
3. OW-HDMA中介分析核心发现应用我们提出的OW-HDMA流程进行核心分析。SIS步骤从48万位点中预筛选出78个候选位点。图9展示了其中相关性最强的前20个位点的热图,证实了数据中存在复杂的共线性结构,这凸显了使用去偏LASSO等高级方法进行估计的必要性。
最终,在严格控制FDR的条件下,只有一个CpG位点(cg07420333)被鉴定为具有显著的中介效应。我们对比了OW和传统IPW两种加权方法的结果(表2)。两者均成功识别出该位点,其中介效应估计值(α×β)分别为0.243和0.269。值得注意的是,基于OW方法估计的系数标准误更小,这表明在本次数据分析中,OW方法可能提供了更稳定、更高效的估计。
表2:显著中介甲基化位点检测结果对比
| | | | | |
|---|
| OW-HDMA (本文方法) | cg07420333 | | | 0.243 | 0.044 |
| | | | | |
注:括号内为标准误。系数α为负表示童年虐待可能降低该位点甲基化水平;系数β为负表示该位点甲基化水平降低与PTSD风险增加相关。
讨论与展望
本研究成功地将重叠加权(OW)整合到高维中介分析(HDMA)框架中,为探索“童年虐待-DNA甲基化-PTSD”这一复杂因果路径提供了稳健的分析工具。我们不仅初步验证了表观遗传机制可能在此关联中扮演中介角色,更重要的是展示了一套能同时攻克“高维”与“混杂”两大方法论堡垒的完整流程。方法学对比显示,OW在本次分析中表现出优于传统IPW的估计稳定性。
发现的显著中介位点cg07420333是一个重要的起点。它如同一枚精确的“分子坐标”,为后续的生物学功能验证(如其调控的基因、影响的下游通路)指明了方向。当然,统计显著性不等同于生物或临床意义,这一发现需要在独立样本和实验模型中得到进一步证实。
本研究的局限也为未来指明了方向。当前模型基于线性假设,未考虑甲基化位点间可能存在的复杂交互作用;样本量相对高维数据而言仍有提升空间。展望未来,随着多组学数据的整合(如甲基化、转录组、蛋白质组),构建更精细、动态的“环境压力-分子网络-疾病表型”图谱,将是揭示精神疾病复杂机制并最终实现精准预防与干预的关键。

本文中分析的完整智能体、数据、代码、文档分享到会员群,扫描下面二维码即可加群!
资料获取
在公众号后台回复“领资料”,可免费获取数据分析、机器学习、深度学习等学习资料。
点击文末“阅读原文”
获取完整智能体、
代码、数据和文档。