1. UHMK1 通过调控 KHK 可变剪接,重塑肝癌细胞代谢与增殖表型
果糖激酶(KHK)是果糖代谢的关键酶,存在 KHK-A 和 KHK-C 两种可变剪接亚型 —— 其中 KHK-A 可活化磷酸戊糖途径、增强细胞抗氧化能力,是驱动肝癌恶性代谢的核心亚型。
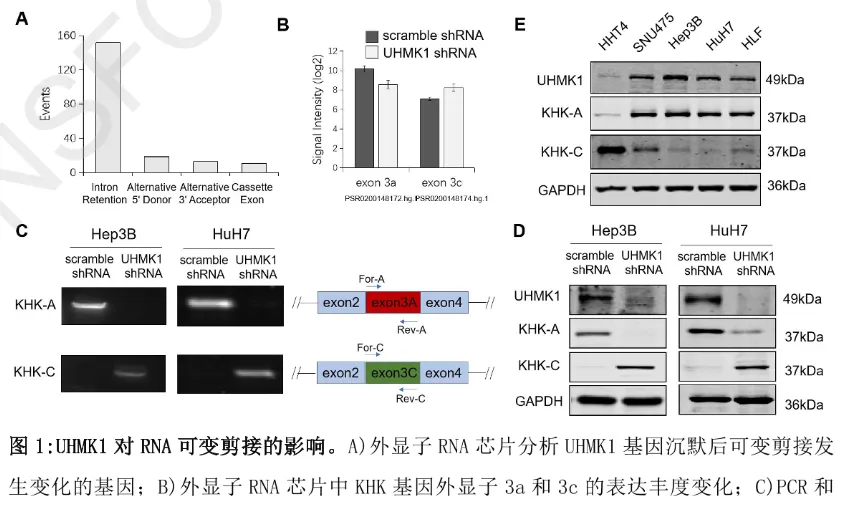
团队通过全基因组外显子芯片分析发现:沉默 UHMK1 可影响 195 个基因的 RNA 可变剪接模式,其中 KHK 的剪接变化最为显著 ——UHMK1 缺失会导致 KHK-A 表达下调、KHK-C 表达上调,且这一趋势在 Hep3B、HuH7 等多株肝癌细胞系中均得到验证(相较于非恶性肝细胞株 HHT4,肝癌细胞中普遍存在 UHMK1 与 KHK-A 高表达、KHK-C 低表达的关联特征)。
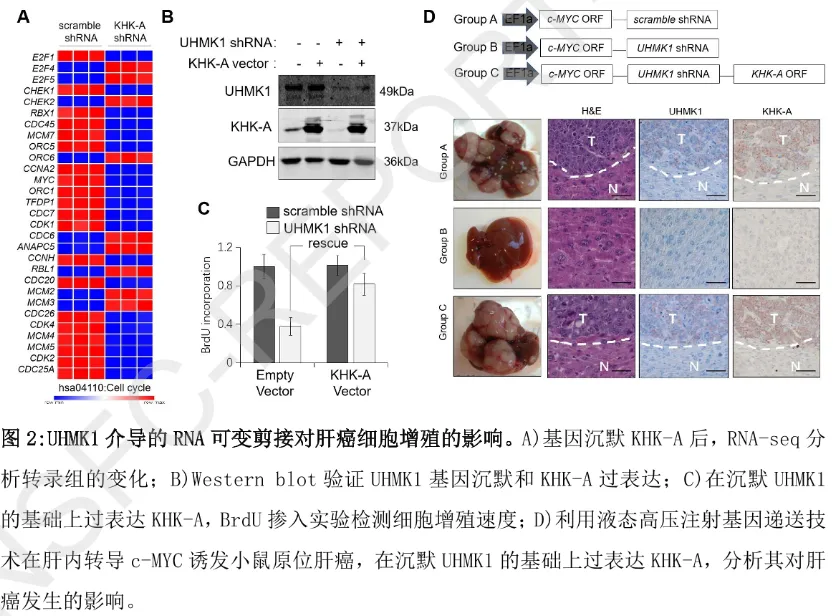
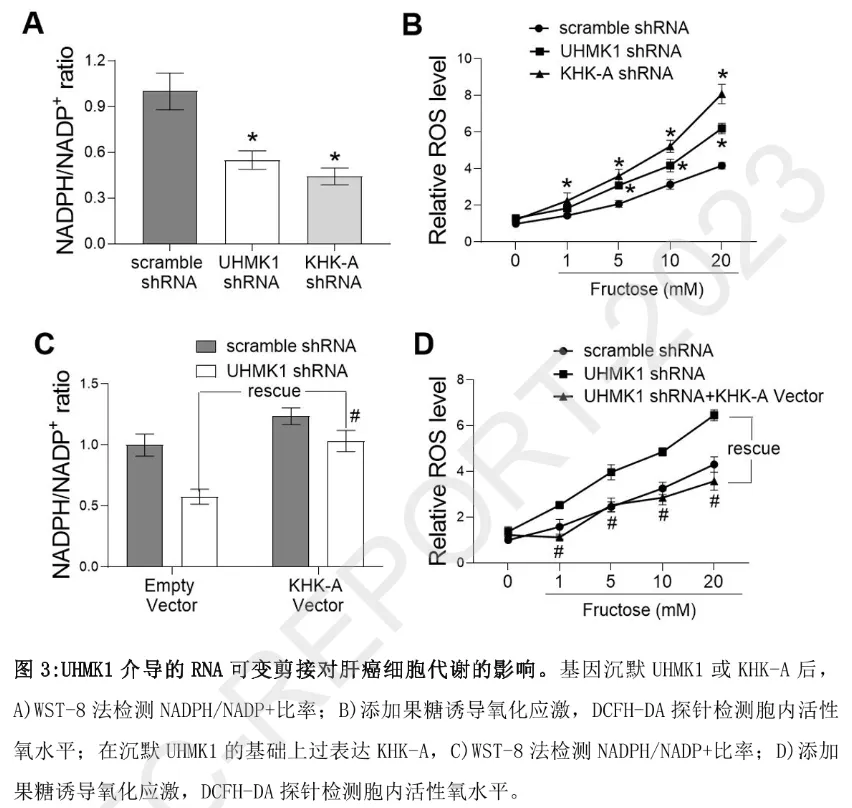
功能实验进一步证实:沉默 UHMK1 或 KHK-A 均会导致肝癌细胞内 NADPH/NADP⁺比率下降(磷酸戊糖途径活性减弱)、活性氧(ROS)清除能力降低,进而抑制细胞增殖;而 “沉默 UHMK1 + 过表达 KHK-A” 的挽救实验可完全逆转上述代谢与增殖缺陷。动物模型(液态高压注射 c-MYC 诱导小鼠原位肝癌)结果显示:沉默 UHMK1 能显著阻断肝癌发生,而恢复 KHK-A 表达可重新驱动肿瘤生长,证实 “UHMK1-KHK 可变剪接” 轴是肝癌体内进展的关键调控通路。
2. UHMK1 通过与剪接因子 hnRNPH1 相互作用,间接调控 KHK 可变剪接
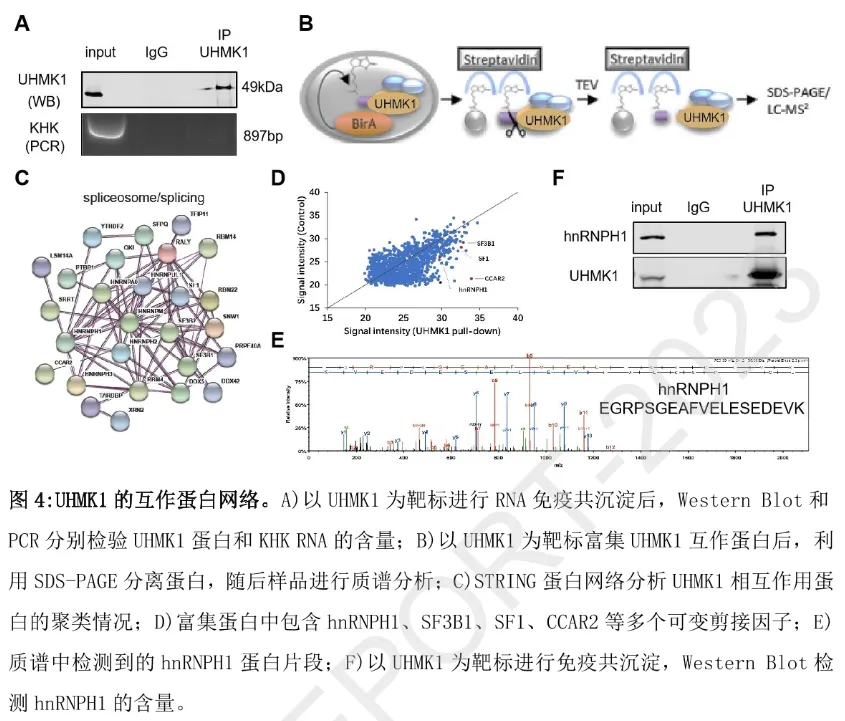
为明确 UHMK1 调控 KHK 剪接的分子媒介,团队通过 RIP 实验发现:UHMK1 并不直接结合 KHK RNA,提示其可能通过剪接复合体间接发挥作用。利用邻近生物素标记(BioID)与蛋白质谱分析,团队从 295 个 UHMK1 互作蛋白中鉴定出 27 个剪接因子,其中 hnRNPH1(异质核糖核蛋白 H1)与 UHMK1 的相互作用经免疫共沉淀(Co-IP)实验证实。
功能验证显示:沉默 hnRNPH1 可模拟 UHMK1 缺失的效应 —— 显著下调 KHK-A 表达、上调 KHK-C 表达,同时导致肝癌细胞增殖抑制、NADPH 生成减少、抗氧化能力减弱;且 RIP 实验证实 hnRNPH1 可直接结合 KHK RNA。上述结果表明,hnRNPH1 是 UHMK1 调控 KHK 可变剪接的关键下游剪接因子。
3. UHMK1 通过激酶结构域介导 hnRNPH1 S310 位点磷酸化,激活其剪接功能
为解析 UHMK1 调控 hnRNPH1 的分子细节,团队构建了激酶结构域失活突变体(UHMK1^K54R)。实验发现:相较于野生型 UHMK1(UHMK1^WT),UHMK1^K54R 完全丧失了与 hnRNPH1 的结合能力,且无法促进 KHK-A 表达;Phos-tag 检测(特异性捕获磷酸化蛋白)进一步证实,UHMK1 可通过激酶活性促进 hnRNPH1 磷酸化。
通过分段克隆与点突变实验,团队明确:hnRNPH1 的 C 端(251-499 位氨基酸)是其与 UHMK1 结合的关键区域,且 UHMK1 介导的磷酸化位点为 hnRNPH1 的 S310 位点(将 S310 突变为丙氨酸 “S310A” 后,hnRNPH1 与 UHMK1 的结合完全消失,且无法调控 KHK 剪接)。
动物模型验证显示:在沉默内源性 hnRNPH1 的小鼠中,过表达野生型 hnRNPH1 可重新驱动肝癌发生,而过表达 S310A 突变型 hnRNPH1 则完全丧失促癌能力,证实 hnRNPH1 S310 磷酸化是肝癌进展的 “不可缺少环节”。