在差异基因分析、多组学数据整合、功能富集分析等生信研究工作中,我们经常需要比较不同实验条件或不同数据库检索结果之间的交集与差异。例如,当你通过两种不同的算法筛选出了各自的差异基因列表,或者需要对比药物靶点数据库与疾病相关基因数据库的重叠情况时,韦恩图(Venn Diagram)就是最直观、最常用的可视化工具。
掌握韦恩图的Python绘制方法,不仅能让你快速完成数据可视化任务,更重要的是理解其背后的代码逻辑和数据结构,这将为后续学习更复杂的生信图表打下基础。本系列课程将采用"完整代码+逐行解析+术语说明"的方式,帮助医务研究人员系统掌握Python科研绘图技能。今天我们从最基础的韦恩图开始。
01 韦恩图的应用场景与数据准备
韦恩图在生信分析中的应用场景非常广泛。在转录组学研究中,你可能需要比较不同处理组的差异表达基因,找出共同上调或下调的基因;在GWAS研究中,可能需要对比不同种族人群的疾病易感位点;在药物研发阶段,需要分析候选药物靶点与已知疾病靶点的重叠情况。这些场景的共同特点是需要可视化展示多个集合之间的交集关系,而韦恩图恰好能够一目了然地呈现这种关系。
从数据格式来看,韦恩图需要的输入数据非常简单,就是两个或三个列表。这些列表可以是基因名称、蛋白质ID、SNP位点编号,或者任何需要比较的元素集合。在Python中,我们通常使用集合(set)这种数据结构来存储这些数据。集合的核心特性是自动去重和支持交集运算,这正是韦恩图计算的基础。
假设你在Excel表格中整理了两组实验数据,实验A发现了BRCA1、TP53、EGFR、MYC这四个基因,实验B发现了TP53、KRAS、EGFR、PIK3CA这四个基因。在Python中,你只需要将它们定义为两个集合即可开始绘图。这种数据准备方式的便捷性,使得韦恩图成为科研工作中最容易上手的可视化工具之一。
02 核心代码逐行解析
下面是绘制韦恩图的完整代码,这是最精简的版本,没有任何冗余内容:
# 导入工具包from matplotlib_venn import venn2import matplotlib.pyplot as plt# 准备数据(两个基因列表)setA = {'BRCA1', 'TP53', 'EGFR', 'MYC'}setB = {'TP53', 'KRAS', 'EGFR', 'PIK3CA'}# 开始画图plt.figure(figsize=(8, 6))venn2([setA, setB], set_labels=('实验A', '实验B'))plt.title('差异基因的韦恩图', fontsize=14)plt.show()
这段代码虽然只有短短几行,但每一行都有其特定的功能和技术含义,我们逐行进行解析:
from matplotlib_venn import venn2
第一行代码是从matplotlib_venn这个第三方库中导入venn2函数。
matplotlib_venn是专门用于绘制韦恩图的工具包,需要通过pip单独安装,它不包含在Python的标准库中。venn2是这个库中专门绘制两圆韦恩图的函数,如果需要绘制三圆韦恩图,则使用venn3函数。这种模块化的设计是Python生态系统的典型特征,不同的可视化需求由不同的专业库来实现。
import matplotlib.pyplot as plt
第二行代码导入的是matplotlib的pyplot模块,这是Python中最基础、最常用的绘图引擎,几乎所有的科研图表都基于它来实现。
这里使用as plt是给这个模块起了一个简称,后续代码中所有的plt都代表matplotlib.pyplot,这是Python编程的惯例写法,可以让代码更加简洁易读。
setA = {'BRCA1', 'TP53', 'EGFR', 'MYC'}setB = {'TP53','KRAS', 'EGFR', 'PIK3CA'}
第三行和第四行代码定义了两个集合setA和setB,使用的是Python的集合数据结构。
注意这里使用的是花括号{},这是集合的标准写法。集合与列表的最大区别在于集合会自动去重,即使你写入了重复的基因名,集合也只会保留一个。更重要的是,集合支持交集、并集、差集等数学运算,这正是韦恩图计算各个区域数值的基础。例如,setA和setB的交集可以通过setA & setB直接计算得到,结果是{'TP53', 'EGFR'},这就是韦恩图中间重叠部分所代表的元素。
plt.figure(figsize=(8, 6))
第五行代码创建了一个绘图画布,figsize参数设置画布的尺寸为8英寸宽、6英寸高。
这个尺寸的设置对于最终图表的清晰度和排版效果至关重要。如果画布太小,图中的文字和数字会显得拥挤,影响可读性;如果画布过大,则可能导致文件体积增大,不利于论文投稿或在线分享(如Nature要求单栏图宽度≤89mm)。8×6英寸是一个比较均衡的尺寸,适合大多数应用场景。
venn2([setA, setB], set_labels=('实验A', '实验B'))
第六行代码是核心绘图命令。
venn2函数接收两个参数,第一个参数是一个列表,列表中包含了要比较的两个集合,注意这里必须用方括号将两个集合包裹起来。第二个参数set_labels是一个元组,用于设置两个圆圈的标签文字。函数在执行时会自动计算setA与setB的交集、setA独有的元素、setB独有的元素,并根据这些数值调整圆圈的大小和重叠程度,最后将结果绘制成韦恩图
plt.title('差异基因的韦恩图', fontsize=14)
第七行代码为图表添加标题,fontsize参数设置字体大小为14磅。在科研论文中,图表必须有清晰的标题来说明图表内容,这是学术规范的基本要求。
第八行代码的作用是将绘制好的图表渲染并显示出来。在Jupyter Notebook环境中,这行代码可以省略,因为Notebook会自动显示图表,但在普通的Python脚本文件(.py文件)中,如果不写这行代码,图表不会显示,只会在后台生成但无法看到。
为了方便理解每行代码的功能,我们用一个简化的对照表来总结:
03 结果解读与数据意义
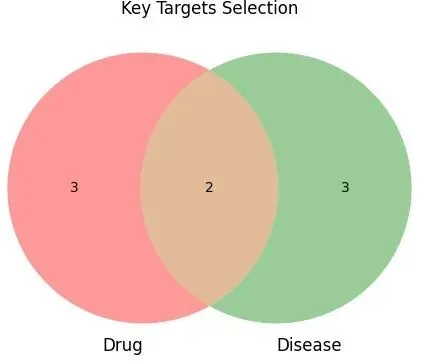
当韦恩图成功绘制后,你会看到两个相互重叠的圆圈,图中会显示三个数字,分别对应三个不同的区域。左侧圆圈中只属于setA的区域显示的数字,代表只在实验A中出现的基因数量;右侧圆圈中只属于setB的区域显示的数字,代表只在实验B中出现的基因数量;而中间重叠部分的数字,则代表同时在两个实验中都出现的基因数量。
图2 韦恩图演示图
在生信分析中,中间重叠部分的基因往往具有最高的研究价值。因为这些基因在两个独立的实验或数据来源中都被识别出来,相当于得到了重复验证,其可靠性远高于只在单一实验中出现的基因。例如,在差异基因分析中,如果一个基因同时被DESeq2和edgeR两种算法识别为差异基因,那么这个基因成为假阳性的可能性就大大降低。在功能富集分析中,如果某个通路同时在KEGG和Reactome数据库中都被富集到,说明该通路的生物学意义更加确凿。
从统计学角度来看,韦恩图的交集大小还可以用来评估两个实验或两种方法的一致性。如果两个本应相似的实验结果交集很小,可能提示实验设计存在问题,或者两种方法的适用条件不同。反之,如果交集过大甚至完全重合,则需要思考两个实验是否存在冗余,或者数据来源是否真正独立。
04 常见问题与解决方案
在实际使用韦恩图的过程中,研究人员经常会遇到一些技术问题,这里总结最常见的四类问题及其解决方案。
第一个常见问题是计算出的交集为0,即韦恩图中间重叠区域显示的数字是0,但你明明知道两个列表中应该有共同元素。这种情况最常见的原因是基因名称的大小写不统一。Python在比较字符串时是严格区分大小写的,EGFR和egfr会被认为是两个完全不同的字符串。如果你的实验A数据来自一个使用全大写基因名的数据库,而实验B数据来自另一个使用首字母大写的数据库,直接比较就会导致交集为空。
解决这个问题的方法是在创建集合时,将所有基因名统一转换为大写或小写。代码如下:
setA = {gene.upper() for gene in ['Egfr', 'Tp53', 'Brca1']}setB = {gene.upper() for gene in ['EGFR', 'KRAS', 'egfr']}这里使用的是集合推导式
(set comprehension)语法,.upper()方法会将字符串中的所有字母转换为大写。经过这样处理后,所有基因名都会统一为大写格式,就能正确计算交集了。
第二个常见问题是运行代码时提示找不到matplotlib_venn模块。这是因为matplotlib_venn不是Python的标准库,需要单独安装。解决方法是在命令行中运行以下命令:
pip install matplotlib-venn
安装完成后,需要重启Python环境或Jupyter Notebook内核,然后重新运行代码即可。
第三个问题是当你需要比较的集合数量超过3个时,韦恩图会变得非常混乱难以阅读。从数学上讲,2个圆的韦恩图有3个区域,3个圆有7个区域,4个圆有15个区域,5个圆有31个区域。当区域数量过多时,圆圈之间的重叠关系会变得极其复杂,不仅难以绘制,读者也很难从图中提取有用信息。
因此,韦恩图只适合用于2到3个集合的比较。如果你需要比较4个或更多集合的交集关系,应该使用UpSet图,这是一种专门为多集合交集可视化设计的图表类型,我们会在后续课程中详细介绍。
第四个问题是中文标题或标签显示为方块乱码。这是因为matplotlib默认使用的字体不支持中文字符。解决方法是在所有绘图代码之前添加以下配置:
import matplotlib.pyplot as pltplt.rcParams['font.sans-serif'] = ['SimHei']plt.rcParams['axes.unicode_minus'] = False
第一行代码将matplotlib的默认无衬线字体设置为黑体(SimHei),这是Windows系统自带的中文字体。如果你使用的是Mac系统,可以将'SimHei'改为'Arial Unicode MS'。第二行代码解决了负号显示异常的问题,因为中文字体对某些特殊符号的支持与英文字体不同。
为了便于快速排查问题,这里提供一个自查清单:
图3 常见问题自查清单
05 Python数据结构术语说明
在韦恩图的代码中,我们使用了集合(set)这种数据结构。为了帮助你更好地理解为什么选择集合而不是列表,这里简要说明Python中三种常用数据结构的区别。
集合(set)是一种无序的、不重复的元素容器,使用花括号{}定义。集合的核心优势是自动去重和支持高效的数学运算,如交集&、并集|、差集-等。在韦恩图的应用场景中,我们需要计算两个基因列表的交集,使用集合可以直接通过setA & setB得到结果,代码简洁且运行效率高。
列表(list)是一种有序的、可重复的元素容器,使用方括号[]定义。列表保留元素的插入顺序,允许通过索引访问特定位置的元素,也允许同一个元素出现多次。如果你的数据需要保持顺序,或者确实需要记录重复元素的出现次数,那么应该使用列表。但对于韦恩图来说,我们只关心某个基因是否出现,不关心它出现了几次,所以集合更合适。
元组(tuple)是一种有序的、不可修改的元素容器,使用圆括号()定义。元组一旦创建就不能增删改其中的元素,这种特性使得元组常用于存储不应该被修改的数据,如函数的返回值、配置参数等。在前面的代码中,set_labels=('实验A', '实验B')使用的就是元组,因为标签一旦设置就不需要改变。
理解这三种数据结构的区别,可以帮助你在编写生信分析代码时选择最合适的数据容器,避免因数据结构选择不当导致的错误或性能问题。
06 思考与讨论
学习完韦恩图的绘制方法后,我给大家留两个思考题,这些问题既能帮助你巩固今天学到的知识,也能引导你思考韦恩图在实际科研中的应用。
第一个问题:如果你将setA和setB设置为完全相同的基因列表,运行代码后韦恩图会呈现什么形态?两个圆圈会如何重叠?这种情况在实际的生信分析中意味着什么?
第二个问题:如果两个实验的结果完全没有交集,韦恩图的中间区域会显示什么数字?从生物学角度,你如何解释两个本应相关的实验却得到了完全不同的基因列表?这种情况下你会如何检查实验设计或数据处理流程?
欢迎在评论区分享你的答案和想法。如果你在实际工作中遇到过有趣的韦恩图应用场景,或者在绘图过程中遇到了本文未涉及的问题,也欢迎留言讨论。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
