使用python读取疑难杂症单细胞数据:GSE135893
- 2026-07-06 03:06:03
python基本部分学完了之后,见专辑《python生信笔记》,就需要开始进行大量的数据分析实战练习进行掌握了。今天来学习一篇顶刊杂志的单细胞数据处理,使用python!
note:这个数据主要来自最近开一个直播进行实战训练,详细情况以及进群方式见:python单细胞数据分析实战直播交流群
整个直播计划见文档:https://www.yuque.com/xinnigegui-uqv7c/nneog4/ecg5ec5hscuiudqq
数据背景
今天分析直播的那篇文献里面里面提到的 Habermann, Kropski et al. 数据,来自文献 10.1126/sciadv.aba1972。文献标题为《Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis》。 2020 年6月8号发表在杂志 Sci Adv. 上。
数据集为:data/12_input_adatas/Habermann_Kropski_2020_pulmonary-fibrosis.h5ad,在文献里面的下载地址为 :https://zenodo.org/records/7227571,在里面的 input_data.tar.xz
文献内容简介
对10例非纤维化对照组和20例PF患者的肺部单细胞悬液进行了单细胞RNA测序。通过对114,396个细胞的分析,鉴定出31个不同的细胞亚群/状态。研究发现,PF患者外周肺组织中出现显著的上皮细胞表型转变,并发现多个先前未被识别的上皮细胞表型,其中包含一种KRT5−/KRT17+的病理型ECM生成上皮细胞群,该细胞群在PF肺组织中高度富集。研究观察到多种成纤维细胞亚型以空间特异性的方式参与ECM扩张。
缩写:肺纤维化(Pulmonary fibrosis、PF)
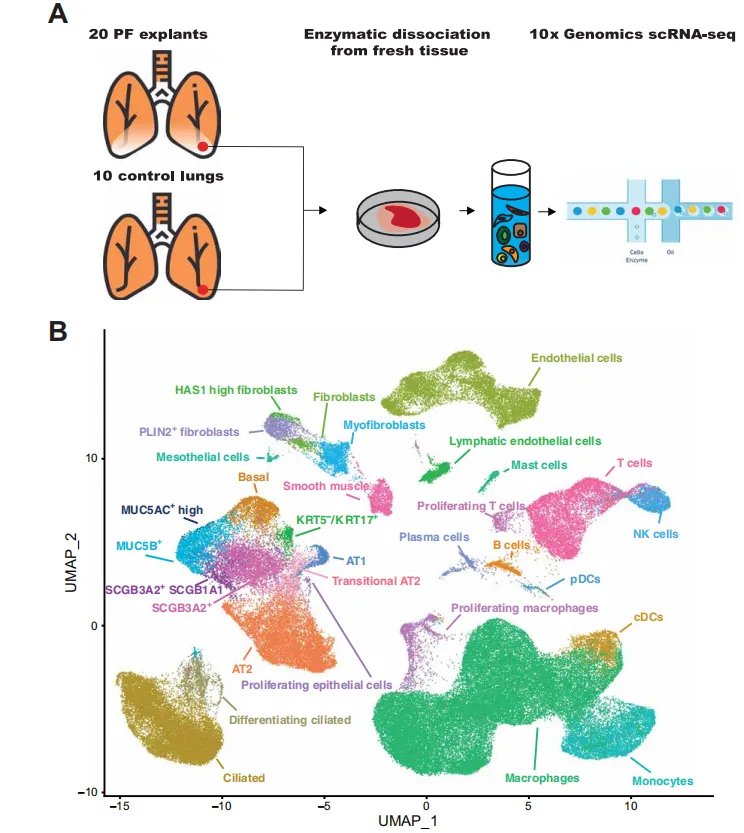
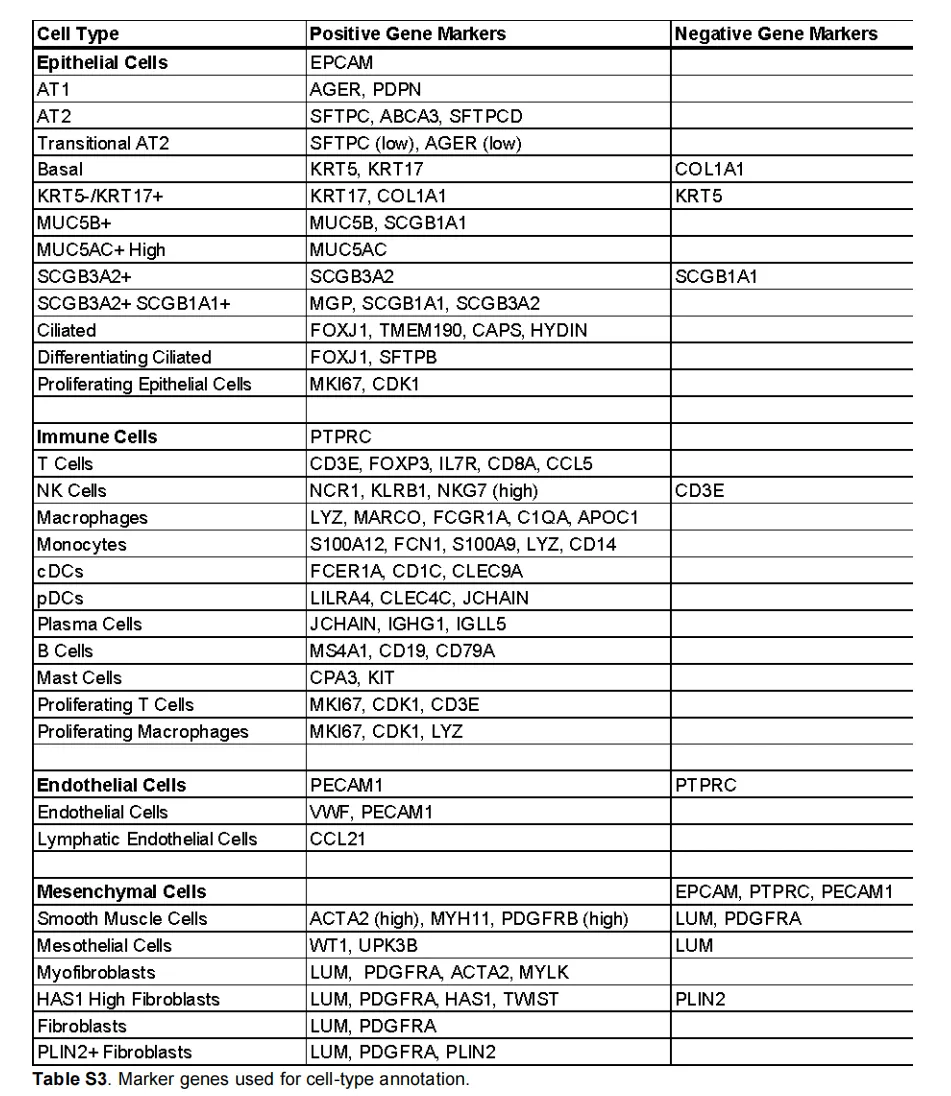
文献的注释方法
大类注释marker基因:
PTPRC+ (immune cells), EPCAM+ (epithelial cells), PECAM1+/PTPRC− (endothelial cells) PTPRC−/EPCAM−/PECAM1− (mesenchymal cells)
亚类型注释:这个marker给的很详细了,用起来!
python读取数据
前面我们学习那个图谱里面的数据都是使用的 作者处理好的h5ad,然后走经典的降维聚类分群,今天我们从 更上游一点的数据开始,锻炼我们的python代码。
1、数据下载
根据这篇文献,我们可以看到作者把数据放到了GEO上面,GSE135893:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135893
提供的几个文件如下:有R格式的rds文件,还有细胞表型信息 metadata,
还有经典的cellranger 三件套:barcodes.tsv.gz、genes.tsv.gz,matrix.mtx.gz
# Seurat object containing all cells and annotations used in analysisGSE135893_ILD_annotated_fullsize.rds.gz 20.9 Gb (ftp)(http) RDS # Metadata file used to annotate sampleGSE135893_IPF_metadata.csv.gz 3.2 Mb (ftp)(http) CSV# matrixGSE135893_barcodes.tsv.gz 914.9 Kb (ftp)(http) TSVGSE135893_genes.tsv.gz 126.6 Kb (ftp)(http) TSVGSE135893_matrix.mtx.gz 1.0 Gb (ftp)(http) MTX# readmeGSE135893_readme_for_ILD_annotated_fullsize.rds.txt 642 b (ftp)(http) TXT把上面的文件下载下来,开始!
2、加载python模块
依然使用我们的老朋友vscode来运行python,小环境为sc2
# %%# %load_ext autoreload# %autoreload 2import scanpy as scimport numpy as npimport pandas as pdimport scipy.sparse as spfrom scanpy_helpers.annotation import AnnotationHelperfrom nxfvars import nxfvarssc.set_figure_params(figsize=(5, 5))3、使用scanpy读取
这里不能用Scanpy的标准函数进行读取,因为 其中的
features.tsv.gz:这个文件只有一列,所有这里直接分开读取,Scanpy期望该文件至少有3列(基因ID、基因符号、特征类型)。
分开读取!这就是难点!
先了解一下 三个文件内容,自己去看一下:
matrix.mtx 文件里面的 33694 220213 338061955 数值,分别是基因数量,细胞数量,以及有表达量的值的数量33694 个基因:zless features.tsv.gz |wc -l220213 个细胞:zless barcodes.tsv.gz |wc -l先读取 barcodes.tsv.gz:220213 个细胞
import osimport pandas as pdfrom scipy.io import mmreadimport anndatacell = pd.read_csv("step10_Habermann_Kropski_2020/GSE135893/input/barcodes.tsv.gz", header=None)cell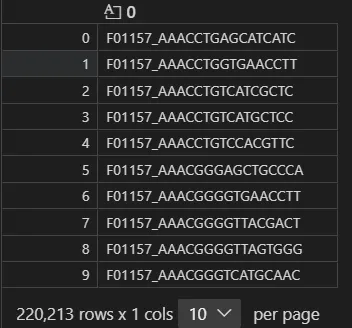
读取 33694 个基因
genes = pd.read_csv("step10_Habermann_Kropski_2020/GSE135893/input/features.tsv.gz", sep='\t', header=None)genes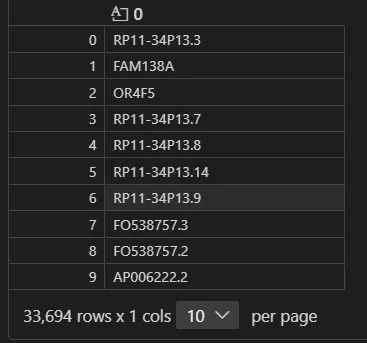
读取mtx
mtx = mmread("step10_Habermann_Kropski_2020/GSE135893/input/matrix.mtx.gz")mtx创建anndata对象
#创建scanpy对象adata = anndata.AnnData(mtx)adata = adata.Tadata.obs.index = pd.Index(cell[0])adata.var.index = pd.Index(genes[0])adata.var['gene_ids']=genes[0].to_list()adata
是不是很简单,后面就跟之前的数据分析一样的套路啦!然后借助上面文献给出的marker,注释看看~
其他类似的数据有:
类似的情况:使用python读取疑难杂症的单细胞数据(GSM7870858)
情况2:scanpy读取数据失败记录:张泽民团队的单细胞数据GSE212890
今天分享到这里~
如果上面的内容对你有用,欢迎一键三连~
转发:
生信入门&数据挖掘线上直播课2026年1月班,你的生物信息学入门课 时隔5年,我们的生信技能树VIP学徒继续招生啦 满足你生信分析计算需求的低价解决方案 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑

