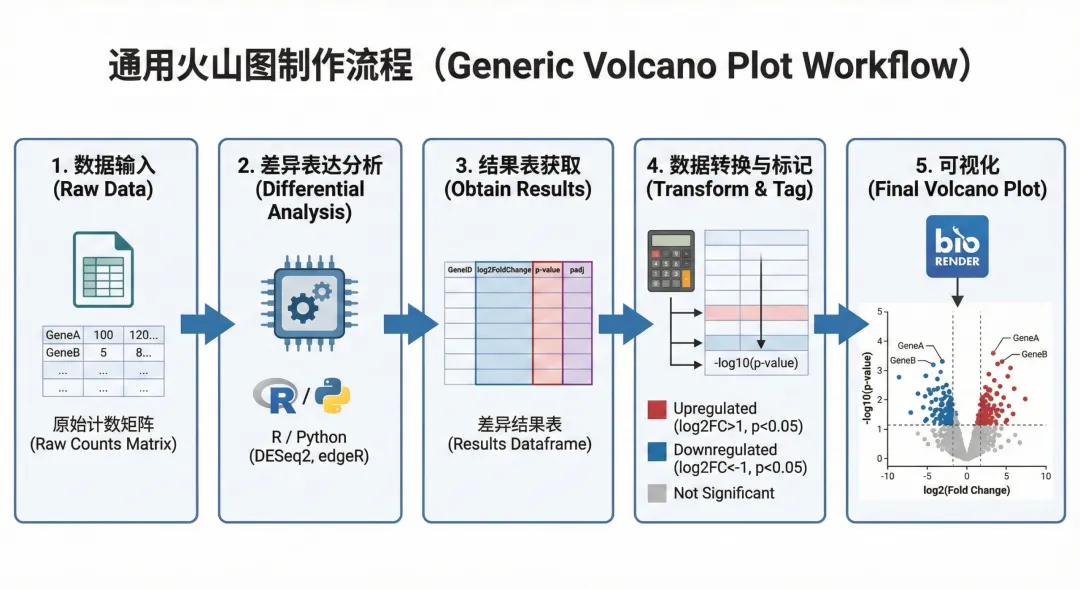
在差异表达基因分析中,我们经常面临这样的问题:如何从成百上千个基因中快速筛选出那些既有显著统计学差异,又具有生物学意义的关键基因?单纯看P值表格效率低下,而火山图(Volcano Plot)正是为解决这一问题而设计的可视化工具。
无论是肿瘤组织与正常组织的对比,用药前后的基因表达变化分析,还是不同疾病亚型的分子特征比较,火山图都能在一张图中同时展示两个维度的信息:横轴呈现基因表达变化的幅度(log2 Fold Change),纵轴展示统计学显著性(-log10 P-value)。这种双重筛选机制让我们能一眼识别出右上角和左上角那些"变化大且显著"的差异基因,大大提升了数据解读效率。
本教程将手把手带你使用Python实现专业级火山图绘制,从数据准备到代码实现,从参数调整到结果解读,帮助你掌握这一差异分析的核心技能。
图1 火山图流程
01 火山图的应用场景与数据准备
火山图专门用于回答一个核心问题:在两组样本的对比中,哪些基因既变化大,又具有统计学显著性?这里的"变化大"指的是表达量的倍数变化(Fold Change),而"统计学显著"则通过P值来衡量。
典型的临床应用场景包括:肿瘤组织与癌旁正常组织的基因表达谱比较,可以帮助我们识别潜在的致癌基因或抑癌基因;治疗前后的转录组对比,能够揭示药物作用的分子机制;不同疾病亚型之间的差异分析,有助于发现亚型特异性的生物标志物。
在开始绘图之前,你需要准备一个包含以下三列核心数据的表格文件:
第一列是Gene,即基因名称,可以是基因符号或ID,这是每个数据点的标识。第二列是log2FC,全称log2(Fold Change),表示基因表达量变化倍数的对数值。这个数值的正负有明确含义:正值表示该基因在实验组中表达上调,负值表示下调。举例来说,log2FC = 2意味着表达量增加了2的2次方即4倍,log2FC = -1表示表达量降低了2的1次方即2倍。第三列是Pvalue,即统计检验得到的P值,数值越小表示差异越显著,通常我们认为P < 0.05具有统计学意义。
此外,有些数据集还会包含padj列,这是经过多重检验校正后的P值,在分析大量基因时更为严格可靠。数据可以保存为CSV格式,这是一种纯文本的逗号分隔文件,Python读取速度快且兼容性好。
02 核心代码逐行解析
下面是一个基于Matplotlib库的完整火山图绘制代码。这段代码涵盖了从数据读取、数值转换、基因分类到图形绘制的全部流程:
import pandas as pdimport matplotlib.pyplot as pltimport numpy as np# 读取数据df = pd.read_csv('diff_genes.csv')# 计算 -log10(Pvalue)df['-log10(Pvalue)'] = -np.log10(df['Pvalue'])# 设置阈值fc_threshold = 1.0 # log2FC的阈值(表示2倍变化)pvalue_threshold = 0.05 # P值阈值# 给每个基因分类(上调/下调/不显著)def classify_gene(row): if row['Pvalue'] < pvalue_threshold and row['log2FC'] > fc_threshold: return 'Up' # 上调 elif row['Pvalue'] < pvalue_threshold and row['log2FC'] < -fc_threshold: return 'Down' # 下调 else: return 'Normal' # 不显著df['Category'] = df.apply(classify_gene, axis=1)# 开始画图plt.figure(figsize=(10, 8))# 分别画三类点colors = {'Up': 'red', 'Down': 'blue', 'Normal': 'gray'}for category, color in colors.items(): subset = df[df['Category'] == category] plt.scatter(subset['log2FC'], subset['-log10(Pvalue)'], c=color, label=category, alpha=0.6, s=20)# 画辅助线(阈值线)plt.axhline(y=-np.log10(pvalue_threshold), color='black', linestyle='--', linewidth=0.8, label='P=0.05')plt.axvline(x=fc_threshold, color='black', linestyle='--', linewidth=0.8)plt.axvline(x=-fc_threshold, color='black', linestyle='--', linewidth=0.8)# 添加标签和标题plt.xlabel('log2(Fold Change)', fontsize=12)plt.ylabel('-log10(P-value)', fontsize=12)plt.title('Volcano Plot of Differential Gene Expression', fontsize=14)plt.legend()plt.grid(alpha=0.3)plt.show()
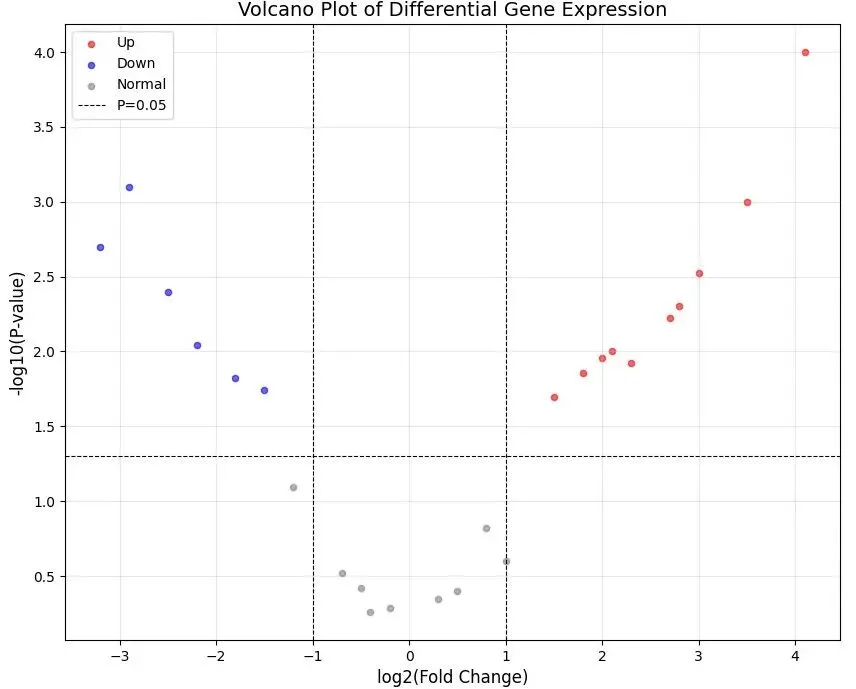
这段代码的执行结果是一张标准的火山图,其中红色点代表上调的显著差异基因,蓝色点代表下调的显著差异基因,灰色点则是变化不显著或变化幅度不够大的基因。图中的虚线标记了我们设定的阈值边界,将整个图形分为不同的区域。
代码逐行解析:
import pandas as pdimport matplotlib.pyplot as pltimport numpy as np
第一:导入必要的库。这三行代码导入了绘制火山图所需的三个核心库。pandas是Python中最常用的数据分析库,专门用于处理表格数据,我们用它来读取CSV文件并进行数据操作。为了方便,我们给它起了个简称pd,后续所有pandas的函数都可以通过pd来调用。matplotlib是Python的标准绘图库,其中的pyplot模块(简称plt)提供了类似MATLAB的绘图接口,用于创建各种统计图表。numpy(简称np)是数值计算库,提供了大量的数学函数,我们主要用它来计算对数值。
df = pd.read_csv('diff_genes.csv')
第二:读取数据文件。这行代码使用pandas的read_csv函数读取CSV格式的数据文件。函数会自动识别文件的第一行作为列名,并将数据加载到一个DataFrame对象中。DataFrame是pandas中"表格"的专业术语,你可以把它理解为Excel表格在Python中的对应物。这里我们将读取的数据赋值给变量df,这是DataFrame的常用缩写,后续所有的数据操作都基于这个df对象。
CSV格式相比Excel文件(.xlsx)的优势在于它是纯文本格式,文件体积小,读取速度快,并且不依赖特定软件,在跨平台数据交换中更为通用。
df['-log10(Pvalue)'] = -np.log10(df['Pvalue'])
第三:P值的对数转换。这是火山图绘制中最关键的一步数据转换。原始的P值范围在0到1之间,且数值越小表示越显著,这与人类的直觉相反(我们更习惯"数值越大越重要")。通过取以10为底的对数并加上负号,我们实现了两个目的。
首先是数值反转。P值为0.05时,-log10(0.05)约等于1.3;P值为0.001时,-log10(0.001)等于3.0;P值为0.00001时,-log10(0.00001)等于5.0。转换后的数值越大,表示原始P值越小,即统计显著性越强,这符合直觉。
其次是尺度拉伸。原始P值在0到1的范围内变化,如果直接作为纵坐标,所有点都会挤在图的底部。经过对数转换后,数值范围扩展到0到正无穷,能够有效拉开不同显著性水平的距离,形成火山图特有的"火山喷发"形状。
fc_threshold = 1.0 # log2FC的阈值(表示2倍变化)pvalue_threshold = 0.05 # P值阈值
第四:设置筛选阈值。这两个阈值是差异基因筛选的标准。fc_threshold设置为1.0,对应的是2倍的表达量变化(因为2的1次方等于2)。如果设置为2.0,则对应4倍变化。这个阈值的选择取决于研究的具体需求,大多数研究使用1.0或1.5作为标准。
pvalue_threshold设置为0.05,这是统计学中广泛接受的显著性水平。在某些要求更严格的研究中,也会使用0.01甚至0.001作为阈值。如果使用了多重检验校正后的padj值,阈值可能需要相应调整。
def classify_gene(row): if row['Pvalue'] < pvalue_threshold and row['log2FC'] > fc_threshold: return 'Up' # 上调 elif row['Pvalue'] < pvalue_threshold and row['log2FC'] < -fc_threshold: return 'Down' # 下调 else: return 'Normal' # 不显著
第五:基因分类函数。这个函数定义了基因分类的逻辑规则。函数接收DataFrame的一行数据作为输入参数(用row表示),然后根据该行的Pvalue和log2FC值进行判断。
分类规则是这样的:如果P值小于阈值(即具有统计显著性)且log2FC大于正阈值,说明该基因在实验组中显著上调,返回'Up';如果P值小于阈值且log2FC小于负阈值,说明该基因显著下调,返回'Down';其他所有情况,包括P值不显著的,或者虽然显著但变化幅度不够大的,都归为'Normal'。
这个三类分类系统是火山图颜色编码的基础,后续我们会给不同类别赋予不同颜色。
df['Category'] = df.apply(classify_gene, axis=1)
第六:这行代码将分类函数应用到DataFrame的每一行。apply函数是pandas中非常强大的工具,它可以对DataFrame的行或列批量应用某个函数。参数axis=1指定按行处理(axis=0是按列处理)。执行后,DataFrame会新增一列名为'Category'的列,每个基因都会被标记为'Up'、'Down'或'Normal'。
plt.figure(figsize=(10, 8))
第七:创建画布并绘制散点。这行代码创建一个新的图形窗口,figsize参数指定图形的大小,单位是英寸,(10, 8)表示宽10英寸、高8英寸。合适的图形尺寸能确保在投影或打印时图表清晰可读。
colors = {'Up': 'red', 'Down': 'blue', 'Normal': 'gray'}for category, color in colors.items(): subset = df[df['Category'] == category] plt.scatter(subset['log2FC'], subset['-log10(Pvalue)'], c=color, label=category, alpha=0.6, s=20)
第八:这段代码是绘图的核心部分。首先定义一个字典colors,建立类别与颜色的对应关系:上调基因用红色,下调基因用蓝色,不显著基因用灰色。这种颜色方案是学术界的通用标准。
然后使用for循环依次处理每个类别。在每次循环中,先通过条件筛选创建一个子集subset,它只包含当前类别的基因。接着调用plt.scatter函数绘制散点图,第一个参数是x坐标(log2FC),第二个参数是y坐标(-log10 Pvalue)。
参数c指定点的颜色,label设置图例标签,alpha设置透明度(0.6表示60%不透明),s设置点的大小。透明度设置很重要,因为在数据点密集的区域,完全不透明的点会相互遮挡,而适当的透明度可以让重叠区域呈现更深的颜色,反映出数据点的密度分布。
这里为什么要分三次绘制而不是一次画完所有点?因为如果一次性绘制,无法给不同类别设置不同颜色。分类绘制是实现颜色编码的必要步骤。
plt.axhline(y=-np.log10(pvalue_threshold), color='black', linestyle='--', linewidth=0.8, label='P=0.05')plt.axvline(x=fc_threshold, color='black', linestyle='--', linewidth=0.8)plt.axvline(x=-fc_threshold, color='black', linestyle='--', linewidth=0.8)
第九:绘制阈值参考线。这三行代码在图上添加阈值参考线。axhline函数绘制水平线,y参数指定线的纵坐标位置,这里是-log10(0.05)约等于1.3,表示P值等于0.05的临界线。axvline函数绘制垂直线,x参数指定线的横坐标位置,我们画了两条垂直线,分别在log2FC等于1和-1的位置。
参数linestyle='--'设置线型为虚线,linewidth=0.8设置线宽,color='black'设置为黑色。这三条参考线将火山图划分为不同区域,帮助快速识别哪些点超过了我们设定的阈值标准。图形被分为九个区域,其中右上角区域(log2FC > 1且P < 0.05)是显著上调基因,左上角区域(log2FC < -1且P < 0.05)是显著下调基因,这两个区域是我们重点关注的对象。
plt.xlabel('log2(Fold Change)', fontsize=12)plt.ylabel('-log10(P-value)', fontsize=12)plt.title('Volcano Plot of Differential Gene Expression', fontsize=14)plt.legend()plt.grid(alpha=0.3)plt.show()
第十:添加图表标注。这几行代码完成图表的标注和显示。xlabel和ylabel分别设置x轴和y轴的标签,fontsize参数控制字体大小。标签需要清晰说明坐标轴的含义,这里使用了标准的学术表述。
title设置图表标题,用稍大的字号(14号)以突出显示。legend函数显示图例,它会根据之前scatter函数中的label参数自动生成,说明不同颜色代表的含义。
grid函数添加网格线,alpha=0.3设置30%的透明度,使网格线不会过于突出而干扰主要数据的观察。最后,show函数将图形显示在屏幕上。
03 火山图的解读方法
完成的火山图呈现出特征性的"火山喷发"形状。图形的中心区域聚集了大量灰色点,这些是表达变化不大或统计学不显著的基因。随着向两侧和上方延伸,点的密度逐渐降低,这部分基因的变化幅度更大或显著性更强。
图形的右上角是红色的上调基因区域,这些基因在实验组中的表达量显著高于对照组,可能代表了致病基因、药物靶点或疾病激活的信号通路。左上角是蓝色的下调基因区域,这些基因的表达被显著抑制,可能是抑癌基因、保护因子或被治疗干预所影响的通路。
在分析时,我们主要关注这两个角落的基因。这些基因不仅通过了统计显著性检验(P < 0.05),还展现出足够大的生物学变化(表达量变化超过2倍)。单纯依靠P值或单纯依靠倍数变化都不够全面,火山图的价值正在于整合了这两个维度的信息。
底部中央的点即使log2FC值较大,但由于P值不显著,说明这种变化可能是随机波动,不具有生物学意义。同样,紧贴水平参考线但接近纵轴的点虽然P值显著,但变化幅度太小,临床意义有限。
04 常见错误及解决方案
在实际绘图过程中,几个常见问题会导致图形异常。
问题一:所有点都挤在图的底部,没有火山形状
这通常是因为纵坐标使用了原始P值而非-log10转换后的值。原始P值的范围是0到1,数值都很小,如果直接作为纵坐标,所有点都会聚集在接近0的位置。正确的做法是使用-np.log10(df['Pvalue'])进行转换,将P值映射到0到正无穷的范围。
错误代码是:
plt.scatter(df['log2FC'], df['Pvalue']) # 错误
正确代码应该是:
plt.scatter(df['log2FC'], -np.log10(df['Pvalue'])) # 正确
问题二:图中所有点都是同一种颜色
这说明没有进行基因分类,或者分类后没有分组绘制。解决方法是确保正确执行了classify_gene函数对基因进行分类,并使用for循环分别绘制三类基因,每类使用不同颜色。如果跳过分类步骤直接用plt.scatter绘制全部数据,就只会得到单色的图。
问题三:水平参考线位置明显错误
阈值线的位置计算错误会导致参考线不在应该的位置。最常见的错误是直接用P值阈值作为y坐标:
正确的写法需要进行对数转换:
plt.axhline(y=-np.log10(0.05)) # 正确,约等于1.3
问题四:数据点过于密集,重叠区域看不清
当基因数量很大时,中心区域的点会高度重叠。可以通过以下方法改善:
首先是调整透明度,将alpha参数从0.6降低到0.3或更低,让重叠的点可以相互透出,密集区域会呈现更深的颜色。
其次是缩小点的大小,将s参数从20减小到10或5,减少点之间的物理重叠。
如果这两种方法还不够,可以添加位置抖动(jitter),给坐标加上微小的随机偏移:
df['log2FC_jitter'] = df['log2FC'] + np.random.normal(0, 0.05, len(df))
然后用jitter后的坐标绘图,这样重叠的点会被轻微分散开。
问题五:想要标注关键基因的名称
在基础火山图上添加基因名标注,可以突出显示最重要的差异基因。实现方法是先筛选出目标基因,然后使用plt.text函数添加文本:
# 选择P值最小的前10个基因top_genes = df.nlargest(10, '-log10(Pvalue)')for idx, row in top_genes.iterrows(): plt.text(row['log2FC'], row['-log10(Pvalue)'], row['Gene'], fontsize=8)
nlargest函数按照指定列的数值大小选出前N行,这里我们选择-log10(Pvalue)最大即P值最小的10个基因。iterrows函数逐行遍历这些基因,plt.text在对应坐标位置添加基因名。可以根据需要调整fontsize控制标注文字的大小。
为了快速排查问题,可以参考以下检查清单:如果点都在底部,检查是否使用了-np.log10(Pvalue);如果没有颜色区分,检查classify_gene函数和for循环;如果阈值线位置不对,检查axhline的y坐标计算;如果点太密集,调整alpha和s参数;如果缺少图例,确认在plt.show()之前调用了plt.legend()。
05 实践与进阶
掌握了基础火山图的绘制后,我们可以通过调整参数来适应不同的研究需求。尝试将阈值修改为fc_threshold=2.0和pvalue_threshold=0.01,观察图形会发生什么变化。更严格的阈值会减少红色和蓝色点的数量,只有那些变化更大、更显著的基因才会被标记出来,这在需要高度特异性筛选时很有用。
另一个实用的练习是为P值最小的前5个基因添加名称标注。使用nlargest函数筛选出这些基因,然后用plt.text函数在对应位置添加基因名,这能让图表更具信息量,在学术汇报中直接指出关键发现。
在下一讲中,我们将学习热图(Heatmap)的绘制方法,它能够同时展示数十甚至上百个基因在多个样本中的表达模式,通过颜色深浅直观呈现基因表达的高低,是差异基因验证和聚类分析的重要工具。
如果你在练习过程中遇到了问题,或者成功绘制出了自己数据的火山图,欢迎在评论区分享你的代码和结果。同时,也期待看到大家对不同阈值设置的尝试效果。让我们通过实践不断提升Python生信绘图的技能。