使用python分析(IF=50.0/Q1)顶刊杂志的单细胞数据(GSE123902)
- 2026-06-27 14:56:18
python基本部分学完了之后,见专辑《python生信笔记》,就需要开始进行大量的数据分析实战练习进行掌握了。今天来学习一篇顶刊杂志的单细胞数据处理,使用python!
note:这个数据主要来自最近开一个直播进行实战训练,详细情况以及进群方式见:python单细胞数据分析实战直播交流群
整个直播计划见文档:https://www.yuque.com/xinnigegui-uqv7c/nneog4/ecg5ec5hscuiudqq
数据背景
今天分析直播的那篇文献里面里面提到的 Laughney, Massague et al. 数据,来自文献 10.1038/s41591-019-0750-6。文献标题为《Regenerative lineages and immune-mediated pruning in lung cancer metastasis》。 2020 年2月10号发表在 nature medicine 杂志(IF=50.0/Q1)上。
数据集为:
data/12_input_adatas/laughney_massague_2020_nsclc.h5ad,在文献里面的下载地址为 :https://zenodo.org/records/7227571,在里面的 input_data.tar.xz
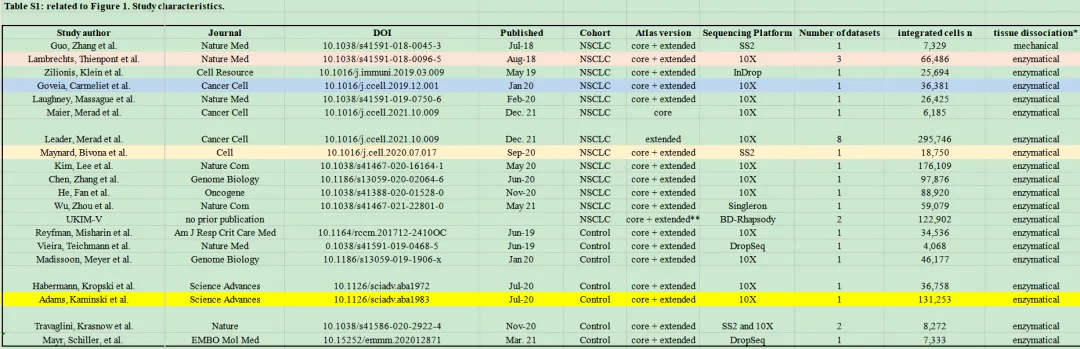
直播文献里面的数据全部在文献的table S1中:
laughney_massague_2020_nsclc 简介
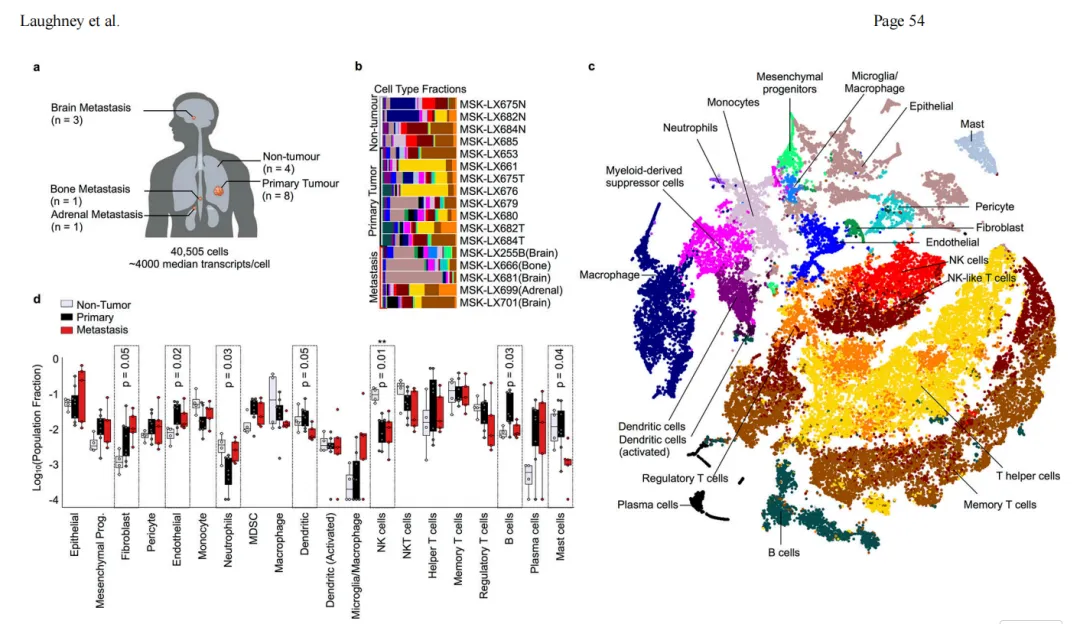
样本信息:对来自 17 个新鲜切除的人类组织样本的 40,505 个单个细胞的转录组进行了分析,包括 normal lung 正常肺(n = 4)、primary lung adenocarcinoma 原发性肺腺癌(LUAD,7个未经治疗 untreated 和1个新辅助化疗后 post neo-adjuvant chemotherapy ),以及来自大脑、骨骼和肾上腺的三个 LUAD 转移瘤(图1a),这些样本涵盖了肿瘤进展的不同阶段。
下面的C图为作者的图谱注释结果:包括癌和非肿瘤上皮,以及肿瘤内的免疫和其他基质细胞类型(n = 40,505个细胞)。
这篇文献我看着很眼熟,原来前面介绍过:
这次直接从作者给的表达矩阵开始(上面的代码都是从作者给的一个奇怪的h5文件开始的,所以那个代码其实不算是很通用。)
单细胞数据上传到了GEO数据库:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123902
17个样本:
GSM3516662 MSK_LX653_PRIMARY_TUMOURGSM3516663 MSK_LX661_PRIMARY_TUMOURGSM3516664 MSK_LX666_METASTASISGSM3516665 MSK_LX675_PRIMARY_TUMOURGSM3516666 MSK_LX675_NORMALGSM3516667 MSK_LX676_PRIMARY_TUMOURGSM3516668 MSK_LX255B_METASTASISGSM3516669 MSK_LX679_PRIMARY_TUMOURGSM3516670 MSK_LX680_PRIMARY_TUMOURGSM3516671 MSK_LX681_METASTASISGSM3516672 MSK_LX682_PRIMARY_TUMOURGSM3516673 MSK_LX682_NORMALGSM3516674 MSK_LX684_PRIMARY_TUMOURGSM3516675 MSK_LX684_NORMALGSM3516676 MSK_LX685_NORMALGSM3516677 MSK_LX699_METASTASISGSM3516678 MSK_LX701_METASTASIS读取参考我们前面给的这个帖子:python版读取不同的单细胞数据格式(单样本与多样本)
step1:加载模块
需要的python模块如下:
# %%# %load_ext autoreload# %autoreload 2import scanpy as scimport numpy as npimport pandas as pdimport scipy.sparse as spfrom scanpy_helpers.annotation import AnnotationHelperfrom nxfvars import nxfvarssc.set_figure_params(figsize=(5, 5))step2:解压GSE123902_RAW.tar
将GSE123902_RAW.tar下载下来,可以很方便的使用 python 解压:
import tarfileimport ostarfile.open("GSE123902_RAW.tar").extractall(path="./GSE123902_RAW")os.listdir('./GSE123902_RAW')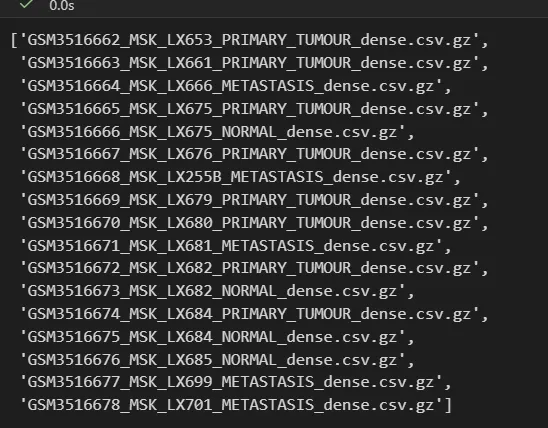
step3:先读取一个样本看看
看下,GSM3516662_MSK_LX653_PRIMARY_TUMOUR_dense.csv.gz,数据为csv格式,第一列为细胞ID,第一行为基因名:
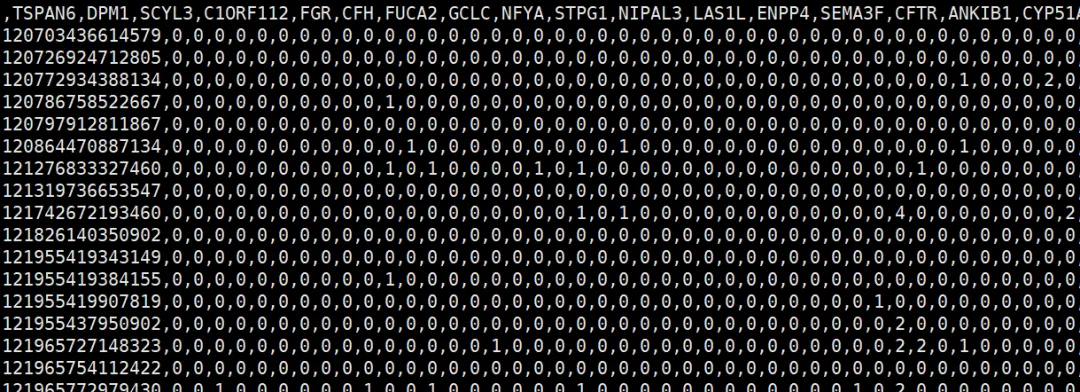
使用 scanpy 读取:
# 读取一个样本的 csv 文件adata = sc.read_csv("./GSE123902_RAW/GSM3516662_MSK_LX653_PRIMARY_TUMOUR_dense.csv.gz",first_column_names=True)adata# 检查结果print(adata.shape)print(adata.obs_names[:5]) # 细胞IDprint(adata.var_names[:5]) # 基因名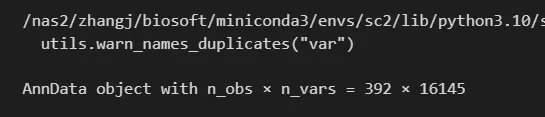
读取完后,adata是一个AnnData对象,n_obs为细胞数,n_vars为基因数。
step4:多个样本读取
一个样本测试成功后,就可以使用 for 循环读取进来啦:
# 多个adata = {}for i in range(len(files)): data = sc.read_csv("./GSE123902_RAW/" + files[i],first_column_names=True) data.var_names_make_unique() name = re.sub(r"_dense.csv.gz*", "", files[i]) adata[name] = dataprint(name)顺利!
然后合并在一起:
# 合并print(adata)adata = sc.concat(adata,label='sampleid')adata.obs_names_make_unique()adata得到的 adata有 42847个细胞:AnnData object with n_obs × n_vars = 42847 × 12604
再看看数据细节:
#表达矩阵里的数值范围np.min(adata.X),np.max(adata.X)#过滤前adata.X.shapeadata[0:6, 'CD3D'].to_df()adata.var.head()adata.var_names[:10]adata.obs.head()step5:画个小提琴看看
先分别计算线粒体以及其他的指标:
# 计算线粒体基因比例adata.var['mt']=adata.var_names.str.startswith('MT-')# 计算核糖体基因比例adata.var['ribo']=adata.var_names.str.match('^RP[SL]')# 计算红血细胞基因比例adata.var['hb']=adata.var_names.str.match('^HB[^(P)]')# 计算sc.pp.calculate_qc_metrics(adata,qc_vars=['mt','ribo','hb'],percent_top=None,log1p=False,inplace=True)# 质控adata.obs.head()
画小提琴:
# 可视化细胞的上述比例情况# jitter:可以设置散点抖动的宽度# rotation:可以调整x轴标签的旋转角度,样本多可以设置90度# size:设置散点的大小from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (11, 10)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="sampleid", jitter=0.4,rotation=90,multi_panel=True,#stripplot=False, # remove the internal dots#inner="box", # adds a boxplot inside violins )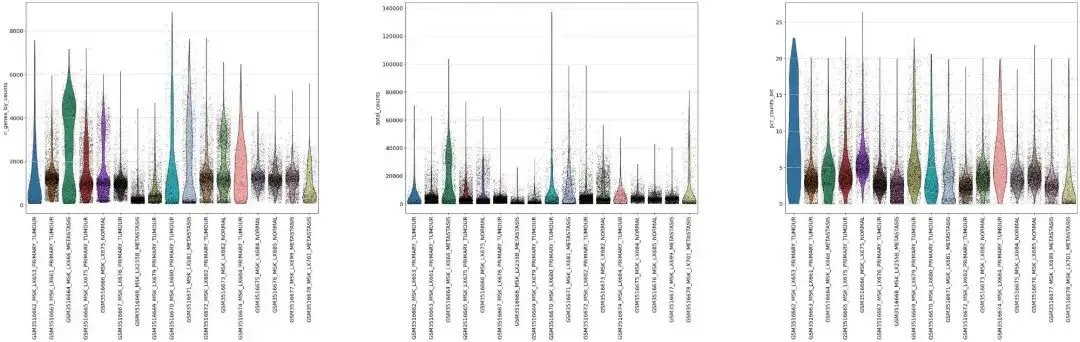
如果你跟了我们这个python单细胞直播系列,现在是不是到了熟练使用python来做单细胞基础分析的程度了呢?
不是的话,来一起学:新叶的微信Biotree123。
今天分享到这里~
如果上面的内容对你有用,欢迎一键三连~
转发:
生信入门&数据挖掘线上直播课2026年1月班,你的生物信息学入门课 时隔5年,我们的生信技能树VIP学徒继续招生啦 满足你生信分析计算需求的低价解决方案 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑

