单细胞技术的飞速发展,让科研人员能在细胞分辨率下解析基因、化学、环境等多种扰动下的分子状态变化,但现有分析方法多聚焦于差异比较、针对特定任务,且缺乏对生物学背景的整合,难以应对高通量扰动数据的规模与复杂性。近日,发表在Nature Methods的研究推出了Pertpy——一款基于Python的模块化单细胞扰动分析框架,作为scverse生态系统的重要成员,它实现了扰动数据的标准化分析、多数据库元数据整合,还兼容主流单细胞分析工具,为扰动组学研究提供了高效、可扩展的全新解决方案。
一、研究背景:单细胞扰动分析的痛点与需求
单细胞扰动实验(如Perturb-seq、CROP-seq)已成为解析细胞响应机制、挖掘药物靶点、探索疾病机制的核心手段,能捕捉基因程序变化、组织间响应差异、细胞互作改变等关键信息。但当前研究面临诸多挑战:
- 现有分析框架(如MUSIC、ScMAGeCK)多聚焦CRISPR扰动,忽略化学、疾病等其他扰动类型,且分析步骤碎片化;
- 缺乏能适配基因组规模数据集的可扩展工具,无法有效整合公共数据库的注释信息;
- 大量工具仅支持R生态,存在维护问题,且难以实现多模态数据的联合分析;
- 扰动数据的低维表征、细胞类型组成变化分析、扰动效应量化等核心需求缺乏统一解决方案。
为解决上述问题,研究团队开发了Pertpy,一款专为单细胞扰动分析设计的端到端框架,实现了从数据加载、元数据注释到扰动效应分析、可视化的全流程覆盖,同时兼顾速度、可扩展性和易用性。
二、核心研究内容:Pertpy框架的设计与功能解析
Pertpy基于Python开发,依托scverse生态实现与scanpy、scvi-tools等主流工具的无缝对接,整合了100余种可组合的分析函数,涵盖扰动数据处理、元数据注释、差异分析、组成分析、扰动空间构建等全流程模块,还通过JAX实现GPU加速,速度显著优于原有工具。其核心设计亮点包括:
- 多类型扰动兼容:支持基因敲除/激活、药物筛选、疾病状态等多种扰动类型,适配单细胞RNA-seq单模态及多模态数据;
- 自动化元数据注释:整合DepMap、GDSC、CMap、PubChem等公共数据库,为细胞系、药物、扰动靶点自动补充注释信息;
- 高效的扰动空间构建:突破传统细胞水平分析,构建扰动空间(单个嵌入代表一种扰动的整体细胞响应),实现扰动效应的直观比较;
- 丰富的量化指标:实现18种以上扰动距离度量(如MSE、Wasserstein距离),支持蒙特卡洛置换检验量化扰动效应;
- 开源可扩展:提供30余个公共扰动数据集的一键加载,预留基础类方便开发者扩展新方法,配套详细教程和文档。
关键Figure解析:Pertpy的功能验证与应用案例
Figure 1 | Pertpy框架的核心模块与分析流程
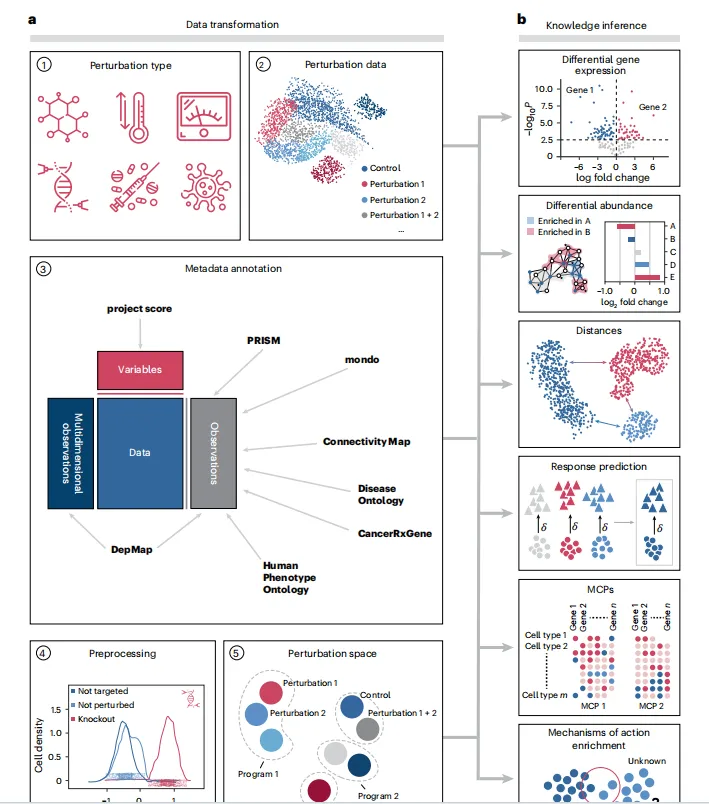
该图全面展示了Pertpy的两大核心分析阶段,清晰呈现了从原始数据到生物学结论的完整流程:
- 左侧(数据处理与扰动空间构建):将基因修饰、化学处理、疾病等单/多模态扰动数据,与Cell Line Ontology、Drug Ontology等本体及DepMap、CMap等数据库整合,完成元数据注释;经预处理去除批次效应、细胞周期等混杂因素后,通过gRNA分配、Mixscape过滤未成功扰动细胞,最终构建有生物学意义的扰动空间,实现从“细胞水平”到“扰动水平”的分析转换。
- 右侧(下游分析模块):根据研究问题灵活选择分析方向,包括差异基因表达分析(支持复杂实验设计)、扰动响应预测、多细胞程序(MCPs)鉴定、扰动间距离计算、药物作用机制富集、细胞类型组成差异分析等,各模块可自由组合构建定制化分析流程。
Figure 2 | 基于Pertpy解析组合CRISPRa扰动的统一扰动空间
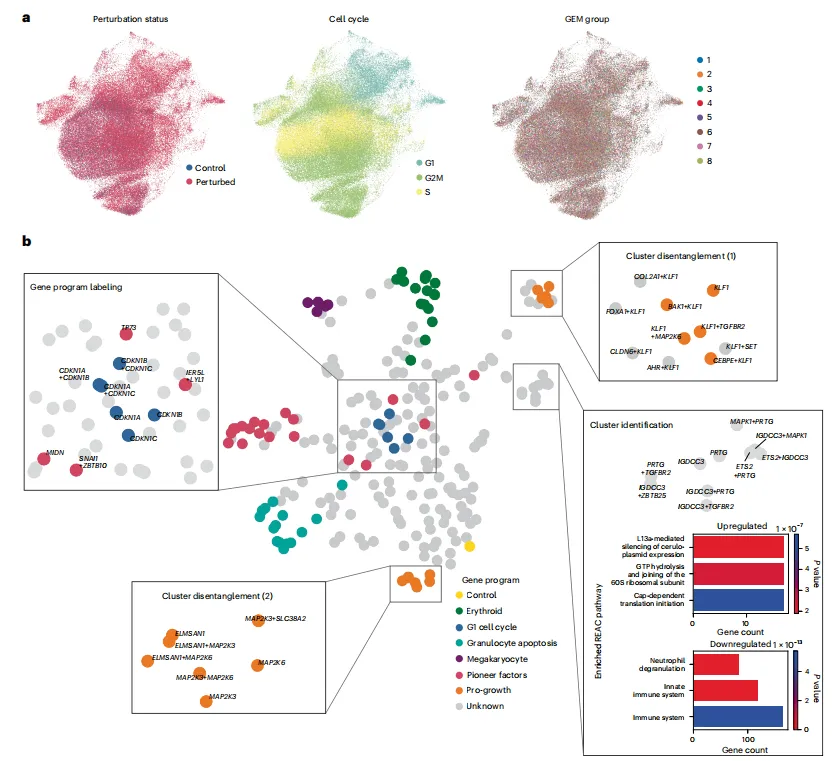
研究以Norman等人的CRISPRa筛选数据集(111,255个K562细胞,287种单基因/基因对扰动)为对象,验证Pertpy构建扰动空间的能力:
- 比较三种预处理策略(近邻对照细胞过滤、同批次对照细胞过滤、无过滤),发现三种策略构建的扰动空间高度相似,说明无过滤策略在该数据集中更适用(CRISPRa无移码突变导致的扰动逃逸问题);
- 扰动空间中,具有相似效应的扰动会聚类在一起,与原始研究的基因程序注释高度一致;通过整合梯度分析,证实分类器能优先识别各基因程序的靶基因(如促生长程序的KLF1),验证了扰动空间的生物学意义;
- 对原始注释的细化发现:TP73(原注释为先锋因子程序)与G1细胞周期扰动聚类(符合TP73调控细胞周期的已知功能);原单一的促生长程序可分为KLF家族靶点和MAPK家族靶点两个亚群;还鉴定出一个未注释的聚类,其扰动显著下调中性粒细胞脱颗粒通路,实现了新基因程序的发现。
该案例证明,Pertpy可通过组合多个模块,从质量控制到扰动空间构建,再到未注释聚类的功能注释,完成全新的分析流程,高效挖掘扰动数据中的生物学规律。
Figure 3 | Pertpy解析药物筛选数据中与存活率相关/无关的响应特征
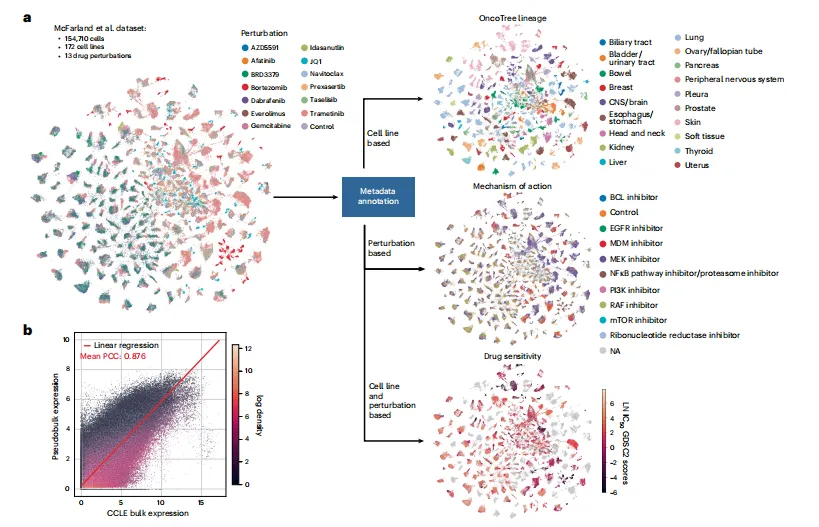
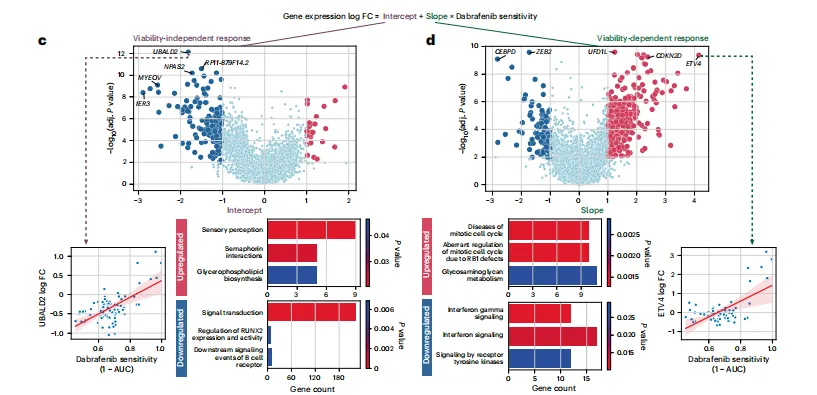
研究利用McFarland等人的MIX-seq药物筛选数据集(154,710个细胞、172种细胞系、13种药物处理),验证Pertpy对大规模化学扰动数据的分析能力:
- 自动化元数据注释:Pertpy一键整合OncoTree、CCLE、DepMap等数据库,为细胞系补充组织来源、癌症类型,为药物补充作用靶点、作用机制,且单细胞表达数据与CCLE批量表达数据的平均皮尔逊相关系数达0.88,验证了数据质量;
- 药物响应的二元分解:以BRAF抑制剂达拉非尼(dabrafenib) 为例,Pertpy通过线性回归将基因表达变化分解为存活率无关响应(截距) 和存活率相关响应(斜率):
- 存活率无关基因:如UBALD2、ETV4,其表达变化与细胞系对药物的敏感性无关,富集于蛋白质翻译通路(为达拉非尼的非经典作用机制提供新线索);
- 存活率相关基因:富集于干扰素信号通路,符合达拉非尼诱导免疫介导的细胞死亡的已知机制;
- 高效复现并扩展了原始研究:Pertpy仅需几步即可完成数据注释、质量控制和统计分析,大幅简化了复杂药物扰动实验的发现流程。
Figure 4 | Pertpy解析三阴性乳腺癌(TNBC)治疗中的复杂扰动效应
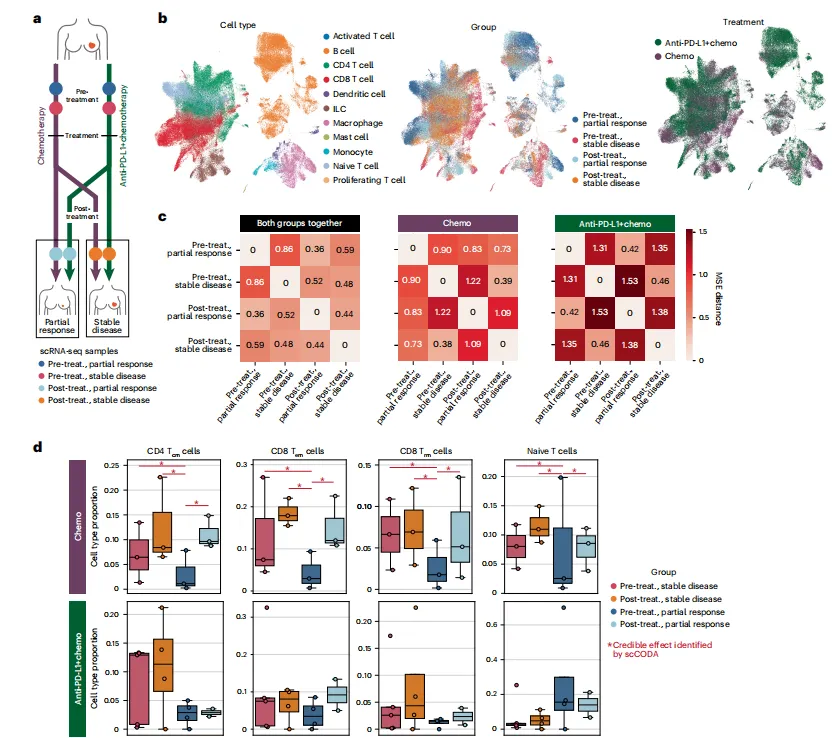
研究以Zhang等人的TNBC数据集(22例患者、新辅助化疗±PD-L1抑制剂治疗)为对象,验证Pertpy在临床组织扰动数据中的应用,解析肿瘤微环境的治疗响应机制:
- 扰动效应量化:计算预处理与后处理样本的MSE距离,发现单独化疗的部分缓解患者的表达谱变化距离显著大于化疗+PD-L1抑制剂组,提示联合治疗的响应强度更低或用于预后更差的病例;
- 细胞类型组成变化:通过scCODA 2.0分析发现,单独化疗组的CD4中枢记忆T细胞、CD8效应记忆T细胞、CD8组织驻留记忆T细胞和初始T细胞的比例发生显著变化,而联合治疗组无明显组成变化,揭示了两种治疗方案对肿瘤免疫微环境的不同影响;
- 多细胞程序(MCPs)鉴定:通过DIALOGUE鉴定出10个MCPs,其中MCP2与治疗响应显著相关,其相关基因富集于热休克蛋白活性和细胞因子信号通路(如IL-7/IL-7R互作),且AP-1复合物核心基因(JUN、FOS、FOSB)在MCP2中显著上调,这些基因可通过调控T细胞行为影响肿瘤治疗效果,为TNBC的治疗靶点挖掘提供了新方向。
三、Pertpy的实验及分析方法流程总结
Pertpy为单细胞扰动分析提供了标准化、可定制的端到端流程,核心步骤可分为数据预处理、核心分析、下游功能挖掘三大阶段,各阶段均配备多种工具和算法,适配不同研究场景,具体流程如下:
阶段1:数据加载与预处理
- 数据加载:支持AnnData/MuData格式,提供30余个公共扰动数据集的一键加载,兼容单/多模态单细胞RNA-seq数据;
- gRNA分配:针对CRISPR扰动,提供阈值法和泊松-高斯混合模型,适配低/高MOI实验设计,准确将gRNA与细胞匹配;
- 质量控制:整合scverse的批次校正方法(如scGen、MNN),去除批次效应、细胞周期等混杂因素;通过Mixscape过滤未成功扰动的细胞,提高分析准确性;
- 元数据注释:自动调用公共数据库API,为细胞系(DepMap/GDSC)、药物(PubChem/ChEMBL)、扰动靶点(CMap)补充注释,实现数据的生物学语境化。
阶段2:核心分析模块(可自由组合)
- 扰动空间构建:提供6种构建方法(如MLP分类器空间、质心空间、伪批量空间),将高维基因表达数据转换为低维扰动嵌入,实现扰动效应的直观比较;
- 差异基因表达分析:提供统一的Formulaic接口,支持PyDESeq2、edgeR、t检验等方法,适配多条件、嵌套比较的复杂实验设计,配套火山图、热图等可视化;
- 有标注组:scCODA 2.0/tascCODA 2.0(贝叶斯方法,分析组成变化,支持细胞类型层级);
- 无标注组/连续比例:Milo(基于k近邻图的差异丰度分析,适配发育等连续过程);
- 多细胞程序(MCPs)鉴定:通过DIALOGUE挖掘组织水平的协同基因表达程序,解析细胞间的功能协作;
- 细胞类型响应排名:Augur(通过机器学习模型预测实验标签,按准确性排名细胞类型的扰动响应);
- 扰动间距离计算:实现18种距离度量(MSE、Wasserstein、欧氏距离等),支持蒙特卡洛置换检验统计验证;
- 因果扰动响应分析:CINEMA-OT(区分混杂变异与扰动效应,实现反事实细胞配对,分析因果效应和协同作用)。
阶段3:下游功能挖掘与可视化
- 基因集富集分析:整合blitzGSEA,基于差异基因或MCP相关基因,挖掘富集的通路、功能注释;
- 作用机制富集:通过Drug2Cell整合药物-靶点-通路信息,解析药物扰动的分子机制;
- 可视化:配套火山图、UMAP、箱线图、热图等多种可视化函数,支持扰动空间、组成变化、MCPs等结果的直观展示;
- 结果验证与扩展:结合公共数据库的注释信息,验证分析结果的生物学意义,挖掘新的靶点和机制。
关键优化与验证
Pertpy对所有集成的方法进行了速度和准确性优化:通过JAX实现GPU加速,运行速度显著优于原始R/Python实现;通过与原始工具的结果对比(如Mixscape、scCODA、Milo),验证了分析结果的一致性(相关系数达0.96以上),确保方法的可靠性。
四、论文结论与展望
核心结论
- Pertpy是首款端到端的Python单细胞扰动分析框架,整合了数据处理、元数据注释、核心分析、可视化的全流程功能,兼容多种扰动类型和数据规模,解决了现有工具碎片化、生态单一、可扩展性差的问题;
- Pertpy通过扰动空间构建、多类型距离度量、自动化元数据整合等核心功能,实现了扰动数据的深度挖掘,能有效发现新的基因程序、解析药物的双重响应特征、揭示临床治疗的免疫微环境变化;
- 作为scverse生态的成员,Pertpy实现了与主流单细胞分析工具的无缝对接,且开源可扩展,为扰动组学研究提供了标准化、高效的分析平台,降低了研究门槛;
- 三个典型应用案例(CRISPRa筛选、药物高通量筛选、TNBC临床治疗)验证了Pertpy在基础研究、药物研发、临床转化中的广泛适用性,能大幅简化分析流程,提升研究效率。
未来展望
- 多模态扰动分析:随着空间转录组、单细胞ATAC-seq等多模态技术的发展,Pertpy将整合空间扰动分析功能(如与Squidpy结合),解析空间维度的细胞响应机制;
- 超大规模数据集适配:通过Dask实现内存外计算,适配数亿级细胞的扰动数据集(如Tahoe-100M),满足基因组规模扰动筛选的分析需求;
- 扰动图谱构建:依托scPerturb、PerturBase等数据集,整合Harmonized的扰动数据,构建单细胞扰动图谱,全面表征不同条件下的细胞诱导状态;
- 基础模型支持:为扰动分析相关的生成式基础模型提供标准化的评估指标和分析工具,推动人工智能在扰动组学中的应用;
- 社区生态建设:通过社区协作持续维护和扩展工具库,整合更多新方法,实现与更多组学分析工具的对接,打造扰动组学研究的一站式平台。
Pertpy的推出,填补了单细胞扰动分析领域的框架空白,为从基础研究到临床转化的全链条扰动组学研究提供了强大工具,有望加速基因功能解析、药物靶点发现、疾病机制研究的进程。
论文基础信息
DOI:10.1038/s41592-025-02909-7主要发表单位:德国慕尼黑亥姆霍兹中心计算生物学研究所、慕尼黑工业大学、美国哈佛医学院、欧洲分子生物学实验室等
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
