把生信流程封装成 Python 包:gffkit 的 PyPI 发布实践
做生信分析时,我们经常会写很多“临时脚本”。
一开始,它们可能只是为了解决某个具体问题:处理某个 GFF 文件、筛选一批基因、修正某个注释格式、补充某类 feature。脚本越写越多之后,就会遇到几个问题:
- • 很难形成一个稳定、可复用、可维护的工具(可能过几个月自己都不知道自己写的是什么鬼玩意了)。
这次我尝试把自己用于 GFF 注释整合的三个 Python 脚本封装成一个软件包,命名为 gffkit,并上传到 PyPI。这样别人就可以通过:
pip install gffkit
直接安装使用
为啥要做gffkit
其实单就注释整合这个问题,已经有一款叫AGAT的软件是能做到的AGAT合并多个注释文件的逻辑如下:
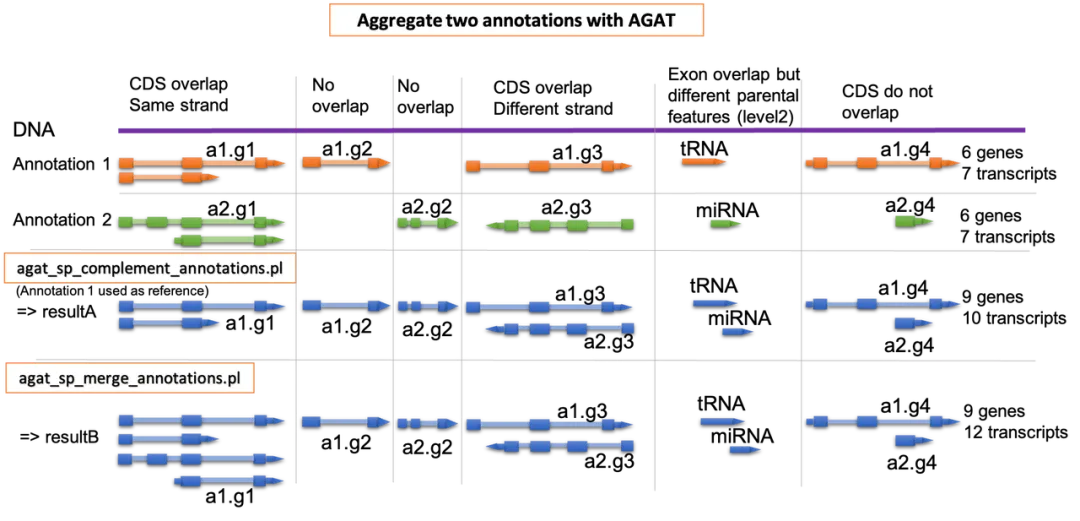 他分两种模式
他分两种模式- 1. 合并模式:完全合并两个注释文件的所有注释信息(来者不拒)
- 2. 互补模式:指定某GFF文件为参考注释,补充参考注释没有而另一个注释文件中独有的注释信息(查缺补漏)
全合并的模式显然有点粗暴,互补模式听上去不错,但它存在一个问题:AGAT的互补模式中会完全以指定的参考注释为基准,例如有两个GFF文件A和B,软件指定了A为参考注释,对某个基因(或者某条染色体的某个区间),A和B的基因结构注释出来的不一样,则结果会完全参照A的结构注释而放弃B的结果,但实际上针对某些特殊的区间可能B的注释效果会更好。
在实际项目中,可能会有这样的一个需求:
我们想在这些区间内以B为参考注释,这些区间以外的用A为参考。
以我的需求为例:
注释 A:EviAnn
这套注释的优点是:
但它也有一个明显问题:
有时会把多个相邻基因错误合并成一个大的 gene locus。
尤其当一个 transcript 出现超长 intron,或者 exon 分布在两个相距很远的区域时,自动注释流程可能会把本应独立的两个基因连成一个基因。
注释 B:ANNEVO
另一套注释来自深度学习流程。它的优点是:
不依赖转录组证据;基因结构更规整;不容易出现多个基因被错误合并的情况。
但它也可能缺少 RNA-seq 证据带来的完整性优势。
所以我希望做的不是简单地“二选一”,而是:
在大多数区域保留注释 A 的优势;在注释 A 出现疑似结构异常的区域,局部切换为注释 B;最后再统一补充 UTR 标签,得到最终版本的 GFF 文件。
这个需求目前AGAT无法实现这就是 gffkit 的设计目标。
gffkit的核心流程
gffkit 目前主要整合了三个脚本
1. detect_bridge_merged_genes.py
这个脚本用于检测注释 A 中潜在的“错误合并基因”。
它关注的典型结构是:
geneX├── transcript1:位于左侧区域├── transcript2:位于左侧区域├── transcript3:位于右侧区域├── transcript4:位于右侧区域└── bridge transcript:一端在左侧,一端在右侧,中间隔着很长距离
脚本的基本逻辑是:
- • 解析 GFF3 中的 gene / transcript / exon / CDS;
- • 对每个 transcript 提取 exon blocks;
- • 判断是否存在超长 intron 或远距离 exon groups;
- • 将这些 transcript 标记为 bridge candidates;
- • 用非 bridge transcript 构建核心 transcript clusters;
- • 如果一个 gene 内存在两个或多个核心簇,并且 bridge transcript 同时连接多个簇,则输出为 suspicious merged gene。
输出结果是:
suspicious.tsv
这个文件记录了疑似假融合基因的坐标区域,后续会作为局部替换区域使用。
2. complement_annotations.py
这个脚本用于GFF 合并。
它的核心思想是:
正常区域:A 作为主参考注释异常区域:B 作为局部主参考注释
也就是说:
在 suspicious.tsv 以外的区域,优先保留 A;在 suspicious.tsv 标记的异常区域,优先使用 B;同时根据 overlap、CDS 重叠等规则补充非冲突的 feature。
3. add_utr.py
第三个脚本用于补充 UTR 标签。
对每个 transcript:
收集 exon 区间;收集 CDS 区间;用 exon 减去 CDS;根据链方向和 CDS 的位置判断:five_prime_UTRthree_prime_UTR
从脚本到软件包:目录结构设计
为了让工具更易用,我把它们整理成了一个 Python 包。
最终项目结构设计如下:
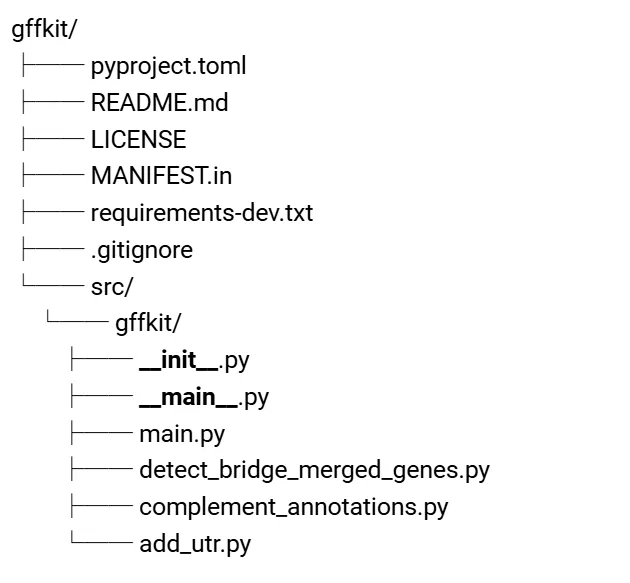
main.py 作为统一入口,用子命令调用三个脚本执行不同的功能
原本用户需要分三步运行:
python detect_bridge_merged_genes.py -i A.gff3 -o suspicious.tsvpython agat_sp_complement_annotations-v3.py \ --ref A.gff3 \ --add B.gff3 \ --swap_region_tsv suspicious.tsv \ --output merged.gff3python add_utr.py \ -i merged.gff3 \ -o final.annotation.withUTR.gff3
封装后可以直接运行:
gffkit integrate \ --annotation-a A.EviAnn.gff3 \ --annotation-b B.ANNEVO.gff3 \ --outdir result \ --prefix sample
输出文件会自动生成:
result/sample.suspicious.tsvresult/sample.merged.gff3result/sample.final.withUTR.gff3
编写配置文件
pyproject.toml
[build-system]requires = ["setuptools>=68", "wheel"]build-backend = "setuptools.build_meta"[project]name = "gffkit"version = "0.1.0"description = "A toolkit for region-aware GFF annotation integration and UTR reconstruction"readme = "README.md"requires-python = ">=3.8"license = { text = "MIT" }authors = [ { name = "Your Name", email = "your_email@example.com" }]keywords = [ "GFF3", "GTF", "genome annotation", "bioinformatics", "UTR", "gene annotation"][project.scripts]gffkit = "gffkit.main:main"gffkit-detect-bridge = "gffkit.detect_bridge_merged_genes:main"gffkit-complement = "gffkit.complement_annotations:main"gffkit-add-utr = "gffkit.add_utr:main"
最重要的是这里面的[project.scripts]gffkit = "gffkit.main:main"
它告诉 Python:
安装这个包以后,创建一个命令行程序 gffkit,并让它调用 gffkit.main 里的 main() 函数。
打包上传PyPI
先注册个账号(https://pypi.org/)
新建个conda环境,安装打包软件所需的依赖
python -m pip install --upgrade build twine
cd 进入你写好的软件目录中(与pyproject.toml同一级)然后
python -m build
这时会生成dist目录
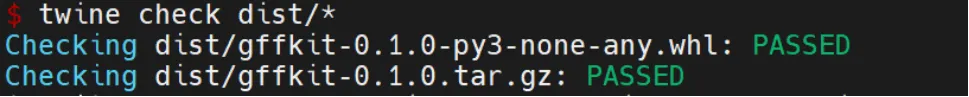
 检查是否成功
检查是否成功twine check dist/*
检查是否成功
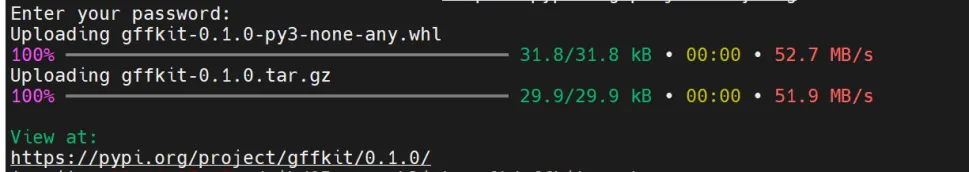
 最后上传
最后上传twine upload dist/*
要输入账号和密码注意这里有个坑username要输入:
__token__
密码就是你登录pypi后网站给的以pypi- 为开头的一长串字符串输入完回车即可
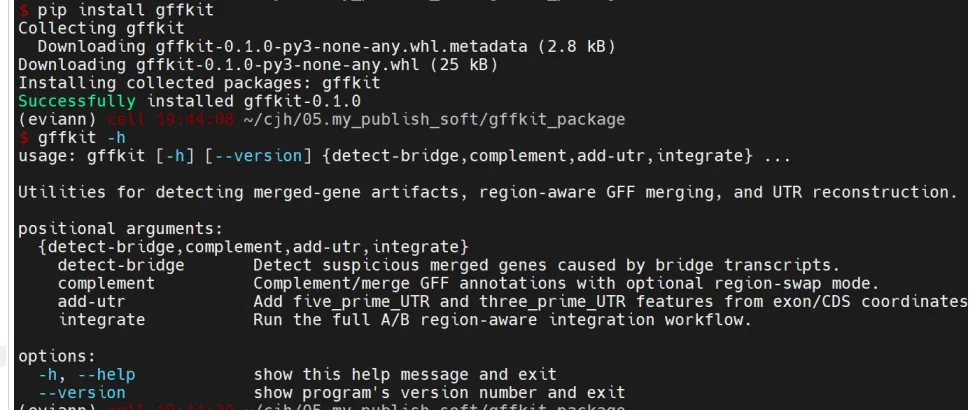
测试一下是能安装成功的
 搞定可以收工了
搞定可以收工了后续如何维护:
- 1. 修改代码例如修改 src/gffkit/*.py
- 2. 修改版本号编辑 pyproject.toml编辑version = "X.X.X"
- 3. 清理旧构建文件rm -rf dist/ build/ .egg-info src/.egg-info src/gffkit.egg-info
- 5. 检查包是否正常twine check dist/*
- 6. 上传 PyPItwine upload dist/*
断更接近一年后的重新更新⁽⁽ ◟(∗ ˊωˋ ∗)◞ ⁾⁾

