单细胞分化轨迹推断领域,Monocle2 是 Trapnell 等在 Nature Biotechnology 2014 提出的经典方法,后续在 Qiu et al. Nature Methods 2017 升级到 v2,核心算法替换为 KDD'15 论文(Mao et al.)提出的 DDRTree —— Discriminative Dimensionality Reduction via learning a Tree。原版 Monocle2 是 R 包, 并且近年已经完全放弃维护,因此用户们要么艰难安装环境,要么转去用 Slingshot、PAGA、Palantir 这些后来者。
Bio-Babel/Monocle2-python 与 Bio-Babel/DDRTree-python 把这两条管线分别从 R 重写成纯 Python:前者负责 Census 计数、density-peak 聚类、order_cells、BEAM 等高层流程,后者负责真正的 backbone —— 同时学习低维嵌入与主图结构的 reversed graph embedding 算法。本推文以 tutorials/V2_1_HSMM.ipynb 为线索,按数据接入 → QC → ordering 基因 → DDRTree → order_cells → 可视化 → BEAM 的次序走完 HSMM myoblast 分化的全流程,并在中段把 DDRTree 的目标函数与迭代步逐行拆开。
总览
Monocle2 解决的问题是把无标签、无时间序列的单细胞表达谱排成一条带分支的连续轨迹,并给每个细胞一个 Pseudotime 数值。做这件事的关键不在差异表达,而在怎么把细胞嵌入到一个低维空间里、并在这个空间里画出一棵主图。这正是 DDRTree 的工作:它在一次优化里同时学三样东西 —— 一个把高维数据压到 2D 的正交投影 W,投影后的细胞坐标 Z,以及一棵显式的主图(K 个节点 Y + MST 边)。Monocle2 拿到 DDRTree 的输出之后,再把每个细胞投到主图最近的边上,在投影点上重建一棵 cell-level MST,从这棵树的几何路径读出 Pseudotime 和分支段标签 State。
Monocle2-python 的 trajectory 部分没有 R 依赖,DDRTree 调用的是同作者维护的纯 Python 包 ddrtree(见 monocle2py/dim_reduction.py:13 的 import ddrtree as _ddrtree),并通过 gold-standard fixture 与 R DDRTree 0.1.6 做数值 parity 测试(tests/test_r_parity.py、tests/test_ddrtree.py)。两个仓库拆开是为了让 DDRTree 这一通用算法能脱离 monocle2 单独使用 —— 任何具有树状内在结构的数据集都可以用它做"嵌入 + 主图"。
数据接入:从 FPKM 到 NB CellDataSet
HSMM 数据集来自 Trapnell 等 Nat Biotechnol 2014 的人骨骼肌成肌细胞分化实验,一共 4 个时间点(0 / 24 / 48 / 72 h)、271 个细胞、47192 个基因,值是 FPKM。Monocle2 的 differential test 与 DDRTree pipeline 都假设负二项(NB)计数模型,而 FPKM 是相对量、有连续小数,不能直接喂进 NB。relative2abs(R 端的 Census 算法)负责把 FPKM 反推回每个细胞的转录本绝对计数:它的核心想法是,在对数 FPKM 的密度估计下,单细胞表达谱在中-低表达区间存在一个由 Poisson 噪声主导的众数,通过对齐这个众数就能恢复总转录本数的尺度。算法实现见 monocle2py/census.py,核心是 bw.nrd0 Silverman 带宽 + KDE 找峰。
import numpy as npimport pandas as pdfrom monocle2py import ( beam, cluster_cells, detect_genes, differential_gene_test, estimate_size_factors, load_hsmm_fpkm, new_cell_dataset, order_cells, plot_cell_clusters, plot_cell_trajectory, plot_genes_branched_heatmap, plot_genes_branched_pseudotime, plot_genes_in_pseudotime, plot_pseudotime_heatmap, plot_rho_delta, reduce_dimension, relative2abs, set_ordering_filter, tobit,)from monocle2py._uns import set_disp_fit_info
fpkm = load_hsmm_fpkm()fpkmfpkm_cds = new_cell_dataset( fpkm.X, pheno_data=fpkm.obs.copy(), feature_data=fpkm.var.copy(), lower_detection_limit=0.1, expression_family=tobit(lower=0.1),)rpc = relative2abs(fpkm_cds, method='num_genes')HSMM = new_cell_dataset( rpc, pheno_data=fpkm.obs.copy(), feature_data=fpkm.var.copy(), lower_detection_limit=0.5,)estimate_size_factors(HSMM)detect_genes(HSMM, min_expr=0.1)
第一次建 CDS 用 tobit family 是因为 relative2abs 内部要做 KDE,需要把 0 当作 left-censored 处理而不是真零;relative2abs 出来的就是 NB 友好的整数计数,第二次建 CDS 用默认的 NB family 即可。estimate_size_factors 给每个细胞算一个 Size_Factor,后续 reduce_dimension 在 norm_method='log' 分支会用它做 library-size 归一化(dim_reduction.py:99-104)。
细胞级 QC
三道 sequential cuts:depth、doublet guard、log10-mean ± 2σ 修剪。
HSMM = HSMM[HSMM.obs['Mapped.Fragments'].astype(float) > 1e6].copy()HSMM.obs['Total_mRNAs'] = np.asarray(HSMM.X.sum(axis=1)).ravel()HSMM = HSMM[HSMM.obs['Total_mRNAs'] < 1e6].copy()log_total = np.log10(HSMM.obs['Total_mRNAs'].to_numpy(float))upper = 10 ** (log_total.mean() + 2 * log_total.std(ddof=1))lower = 10 ** (log_total.mean() - 2 * log_total.std(ddof=1))keep = (HSMM.obs['Total_mRNAs'] > lower) & (HSMM.obs['Total_mRNAs'] < upper)HSMM = HSMM[keep].copy()detect_genes(HSMM, min_expr=0.1)print(f'After QC: cells = {HSMM.n_obs} genes = {HSMM.n_vars}')
271 个细胞过完 QC 剩 262 个,Total_mRNAs 窗口约 [19794, 39569]。两端各 2σ 的截断比 scanpy 默认更宽容一点,因为 monocle2 的下游 NB 模型对极端值不敏感,但 trajectory 几何对孤立点很敏感(它们会被 DDRTree 误当成树的端点),所以先剔掉。
提取 Myoblast 亚群
is_myo = (HSMM.obs['CellType'] == 'Myoblast').to_numpy()HSMM_myo = HSMM[is_myo].copy()estimate_size_factors(HSMM_myo)detect_genes(HSMM_myo, min_expr=0.1)expressed = HSMM_myo.var_names[HSMM_myo.var['num_cells_expressed'] >= 10].tolist()
CellType 是预先标注好的(基于 MYF5+ → Myoblast、ANPEP+ → Fibroblast)。剔掉 Fibroblast 的原因是它和 Myoblast 在表达谱上有大段独立的特征基因 —— 不剔掉的话,DDRTree 学到的主图会被两个亚群之间的鸿沟主导,真正想看的成肌分化轴反而被压缩成一小段。剩下 85 个 Myoblast、约 10554 个表达基因。
选 ordering 基因:density-peak 聚类 + ~Cluster DEG
DDRTree 对 ordering 基因(进入嵌入的特征子集)很敏感:基因选错了,树形也会错。Monocle2 推荐的做法是先做无监督聚类、再用差异基因选择,而不是直接用 ~Media 这种 metadata-driven DEG —— 后者会得到一条过于线性的轨迹,看不出 cycling myoblast / myotube / reserve cell 三支的分支结构。
无监督聚类这里用的是 density-peak(Rodriguez & Laio Science 2014),实现在 monocle2py/clustering.py。它的核心是为每个点算两个量:局部密度 ρ(高斯核 ρᵢ = Σⱼ≠ᵢ exp(-(dᵢⱼ/dc)²),dc 经过二分搜索使平均近邻率落在 1–2% 区间)和到更高密度点的距离 δ(δᵢ = min{dᵢⱼ : ρⱼ > ρᵢ})。簇心是 ρ 与 δ 都偏大的点 —— 既密集又孤立。和 KMeans 比,density-peak 不需要预设簇数;和 Louvain 比,它对 perplexity / resolution 这种隐参数不敏感。代价是 O(N²) 距离矩阵,所以 monocle2 把它放在 tSNE 降到 3 维之后再做。
HSMM_myo.var['use_for_ordering'] = ( HSMM_myo.var['num_cells_expressed'] > 0.05 * HSMM_myo.n_obs)reduce_dimension( HSMM_myo, max_components=2, num_dim=3, norm_method='log', reduction_method='tSNE', perplexity=10, verbose=False,)cluster_cells(HSMM_myo, gaussian=True)
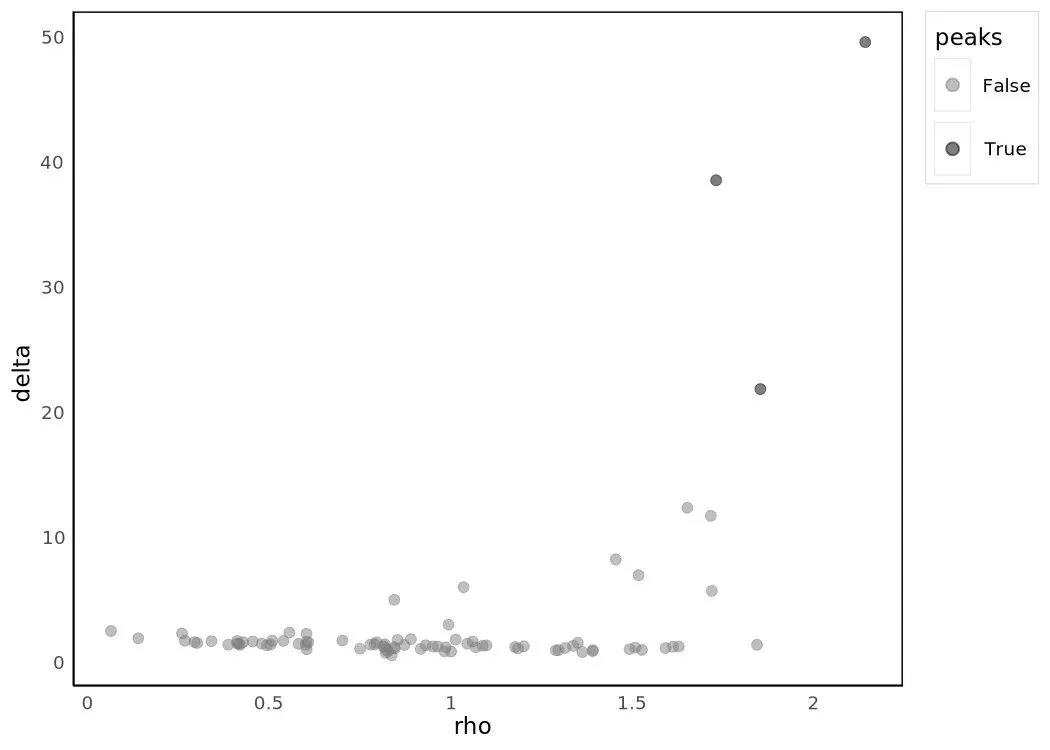
p = plot_rho_delta(HSMM_myo)p
plot_rho_delta 画的就是每个点 (ρ, δ) 的散点 —— 簇心是右上角那几个孤立点。算法据此在 ρ–δ 空间挑簇心,再把每个非簇心点关联到"距离最近的、密度比自己更高的点"所在的簇,从而拿到全部 cluster 标签。
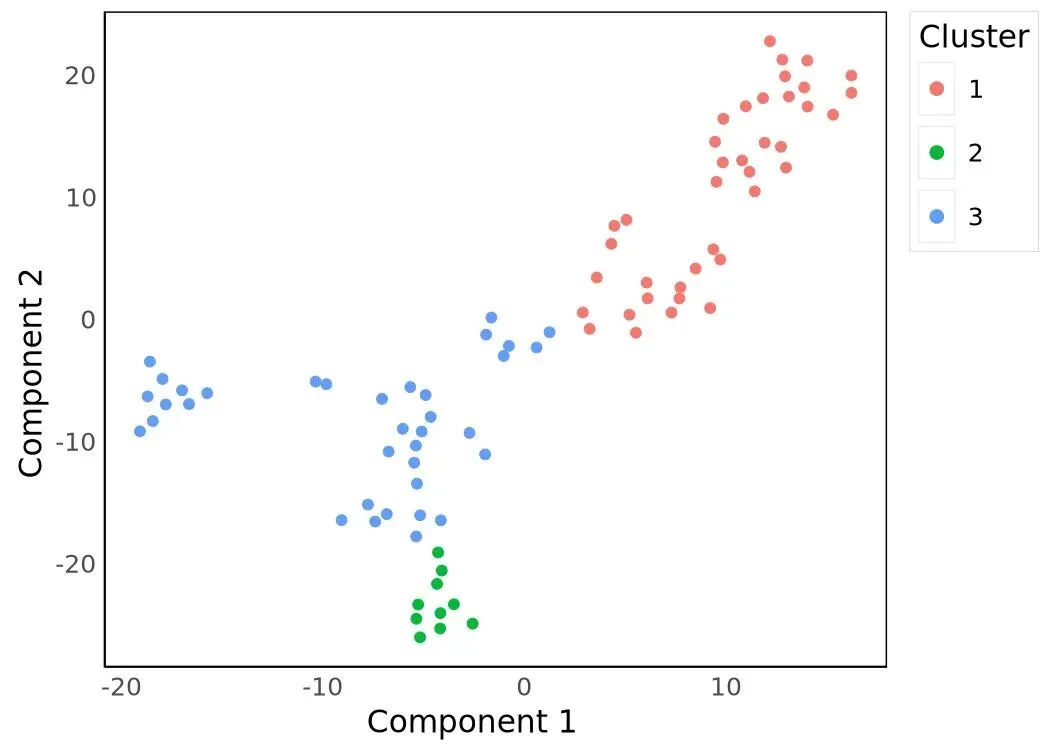
plot_cell_clusters(HSMM_myo)
挑出 3 个 cluster 后,在 panel 上跑 ~Cluster 差异表达检验,取 qval 最小的 1000 个基因作为 ordering filter。这一步把"被 cluster 分布解释的基因"留下来作为 DDRTree 的输入特征:
panel = HSMM_myo[:, [g for g in expressed if g in HSMM_myo.var_names][:200]].copy()panel.uns['monocle2'] = dict(HSMM_myo.uns['monocle2'])def _disp_func(q): return 0.05 + 1.0 / np.asarray(q, dtype=float)set_disp_fit_info( panel, {'disp_func': _disp_func, 'coefficients': {'asymptDisp': 0.05, 'extraPois': 1.0}, 'disp_table': pd.DataFrame()}, name='blind',)clust_DEG = differential_gene_test(panel, '~Cluster', '~1')ordering_genes = clust_DEG.sort_values('qval').index[:1000].tolist()set_ordering_filter(HSMM_myo, ordering_genes)
differential_gene_test('~Cluster', '~1') 内部是个 NB GLM 的 likelihood-ratio test:full 模型 μᵢ = exp(β₀ + βcluster·xᵢ + offset(log Sᵢ)),reduced 模型只有 β₀ + offset。Sᵢ 是 Size_Factor,offset 起到 library-size 归一化的作用。LRT 统计量 2(ll_full − ll_reduced) ~ χ²(df = K_cluster − 1),Benjamini–Hochberg 校正后取 qval。
DDRTree 算法核心
下面进入文章重头戏。reduce_dimension(HSMM_myo, reduction_method='DDRTree') 这一行调用看着平淡,但它把 ordering 基因集打包成一个 D × N 矩阵 X(D = 基因数 = 200,N = 细胞数 = 85)塞给 DDRTree,在大约 10 次迭代里同时学出一个 2D 嵌入和一棵主图。要看懂它学了什么,需要把目标函数和迭代步逐行讲清楚。
5.1 问题设定:reversed graph embedding
传统 graph embedding 是"先有图、再嵌入" —— 拿到一个由 KNN / SNN / 物种树等先验给定的图 G,目标是把节点放到低维空间使图距离尽量被欧氏距离保留(LLE、Laplacian Eigenmaps、UMAP 都在这一脉)。DDRTree 做的事情是反过来:先验只有数据矩阵 X,图本身要从数据里学,并且嵌入与图的学习是同一个优化问题。Mao et al. 把这个范式叫 reversed graph embedding。
DDRTree 一次同时估计 5 个变量:
W ∈ R^{D×d} —— 把数据从 D 维投到 d 维(通常 d=2)的正交基,列是 d 个主向量。Z ∈ R^{d×N}Y ∈ R^{d×K} —— 主图的 K 个节点(质心)在低维空间里的坐标。K 由启发式 cal_ncenter(n_cells) 选,公式 K = round(2 × 100 × ln(N) / (ln(N) + ln(100)))(dim_reduction.py:33-48):85 个细胞 → K ≈ 90,500 个细胞 → K ≈ 152,稳定增长但放缓。B ∈ {0, 1}^{K×K} —— 主图的邻接矩阵,且每次迭代都被强制约束为 K 个节点上的 minimum spanning tree(MST)。R ∈ R^{N×K} —— 软分配矩阵,Rᵢⱼ 是细胞 i 被分到节点 j 的概率,行和为 1。
这 5 个量里只有 X 是给定的;其余全要从 X 反向学出来。
5.2 三项目标函数
DDRTree 把 5 个变量塞进一个目标函数(这里用 Frobenius 范数书写,与 Mao 等 KDD'15 一致):
min_{W, Z, Y, B, R} ‖X − W Z‖²_F + λ · tr(Y L Yᵀ) + γ · Σᵢ σ log Σⱼ exp(-‖zᵢ − yⱼ‖²/σ)
约束:Wᵀ W = I_d,B 是 Y 节点上的 MST,R 行和为 1。三项的物理含义如下。
重构项 ‖X − WZ‖²_F —— 经典 PCA-style 项,要求 Z 通过线性投影 W 还能重建 X。它把"低维空间忠实于原始数据"这件事写进目标。
Laplacian 项 λ tr(Y L Yᵀ) —— L = diag(B·1) − B 是 MST B 上的 graph Laplacian。展开有 tr(Y L Yᵀ) = Σ_(i,j)∈edges ‖yᵢ − yⱼ‖²,即 MST 所有边长的平方和。最小化这一项等价于让主图节点之间互相靠拢、让 MST 总长度短 —— 但因为 MST 本身是用 Y 当前位置上的两两距离构造的,优化时这一项会同时驱动 Y 节点排成尽量短的连通骨架,而骨架在数据真正有分支结构时就会自然分叉,而不是塌成一条线。λ 默认取 5N(_core.py:256-257),意思是平均每个细胞贡献 5 单位的"平滑骨架"惩罚。
软聚类项 γ Σᵢ σ log Σⱼ exp(-‖zᵢ−yⱼ‖²/σ) —— 这是 logsumexp 形式的 entropy-regularized k-means。回忆当 σ → 0 时,σ log Σⱼ exp(-eⱼ/σ) → -minⱼ eⱼ,整项退化成 -γ Σᵢ minⱼ ‖zᵢ−yⱼ‖² 即硬 kmeans;σ 适中时变成"细胞 i 的软分配"。注意整项前面的符号:展开后这一项要被最小化,等价于让每个 zᵢ 至少接近某个 yⱼ。γ=10、σ=1e-3 是 monocle2 默认。这一项把"细胞被吸附到主图最近的节点"写进目标。
把三项凑在一起:第 1 项让 Z 忠实于 X,第 2 项让主图 Y 顺滑且短,第 3 项让 Z 紧贴 Y。三者同时优化的结果就是:Z 既保留数据主要变异,又坐落在一棵"贯穿数据骨干"的树周围。分支结构是这三项的自然平衡产物,不是事后启发式拼出来的。
5.3 迭代步:逐行对应代码
DDRTree 的优化是块坐标下降(block coordinate descent):每次循环依次更新 B → R → 监控目标 → 收敛检查 → W, Z, Y。代码在 ddrtree/src/ddrtree/_core.py:_ddrtree_reduce_dim,主循环 296–402 行,我把每步的更新公式与代码对应列出来。
(1) 更新 B —— MST over current Y centers_core.py:301-314
distsqMU = sq_dist(Y, Y) # K×K 平方欧氏距离mst = _prim_mst(distsqMU) # numpy backend 默认 dense PrimB = (mst > 0).astype(float)
K 个 Y 节点的两两平方欧氏距离构造一个完全图,在上面跑 Prim's 算法得到 MST 邻接矩阵 B。Numpy backend 用 O(K²) 经典 dense Prim 从顶点 0 开始(对应 R src/DDRTree.cpp 里调用的 Boost.Graph prim_minimum_spanning_tree);torch backend 默认用 parallel Borůvka,在 GPU 上 O(log K) 轮就能并行做完。两者在边权唯一时给同一棵树,只在 tie-breaking 上可能有别。
(2) 更新 R —— 软分配 softmax_core.py:319-325
distZY = sq_dist(Z, Y) # N×Ktmp = exp(-(distZY − rowmin) / σ) # logsumexp 数值稳定版R = tmp / rowsum(tmp) # 行归一化
R 的每一行是细胞 i 对 K 个 Y 节点的软分配概率。σ 是温度:小 σ 让 R 接近 one-hot(硬分配),大 σ 让 R 接近均匀。减去 rowmin 是 logsumexp trick,避免 exp 溢出。
(3) 监控目标值 + 收敛检查_core.py:330-355
按 5.2 的目标函数计算当前 obj2,与上一轮做相对差 delta = |obj₂ − obj₁| / |obj₁|,小于 tol=1e-3 就早停。最大迭代 max_iter=10(monocle2 调用时传)。
(4) 联合更新 W 与 Z —— 广义 PCA_core.py:357-392
这一步推导比较绕,但代码很短。先解一个 K×K 的辅助线性系统:
M = (Γ + (λ/γ)·L) · (γ+1)/γ − Rᵀ R # K×Ksol = solve(M, Rᵀ) # K×N (Cholesky 优先,失败回退 LU)Q_eff = (X + X·solᵀ·Rᵀ) / (γ+1) # D×N
其中 Γ = diag(colSums(R))。然后 W 的更新是:tmp1 = Q_eff · Xᵀ,取对称化矩阵 (tmp1 + tmp1ᵀ)/2 的 top-d 个特征向量(代码用 pca_projection,内部调 scipy.sparse.linalg.svds,Lanczos 系列,模仿 R 的 irlba)。直觉:Q_eff 是把 X 用当前的软分配 R 与 Y、L 做了一次"被树骨架重新加权"的版本;它的协方差矩阵的 top-d 特征向量给出了"既要还原 X、又要让投影点贴近树"的最佳 d 维子空间。然后:
W = top_d_eigvecs( (tmp1 + tmp1ᵀ)/2 ) # D×dZ = Wᵀ · Q_eff # d×N
这一段把 PCA 推广成"reversed graph embedding 的 PCA"—— 普通 PCA 只看 X Xᵀ,DDRTree 看的是被软分配和树 Laplacian 共同重塑过的 Q_eff Xᵀ。
(5) 更新 Y —— 解一个 PSD 线性系统_core.py:395-402
Y_lhs = (λ/γ)·L + Γ # K×K, PSDLc = cholesky(Y_lhs)Y = solve_chol(Lc, (Z R)ᵀ).ᵀ # d×K
(λ/γ)L + Γ 是图 Laplacian + 非负对角,构造上严格 PSD,Cholesky 必成功。这一行的几何含义:在固定的 Z 和软分配 R 下,Y 的最优解是把 Z 用 R 加权做"软质心",再被树平滑 λL/γ 沿边拉直 —— 二者通过 Cholesky 一次性解出。
5 步循环 max_iter=10 次,目标值单调下降到 plateau,得到 (W, Z, Y, B)。代码末尾把 K×K 的 MST 嵌入到 N×N 稀疏矩阵 stree(只填左上 K×K 块),作为对 R 端 DDRTree 返回 list 的形状对齐(_core.py:404-412)。
至此 DDRTree 算法解读完成。下一步看 monocle2 怎么把这个输出对接到 trajectory。
reduce_dimension + order_cells
reduce_dimension( HSMM_myo, max_components=2, reduction_method='DDRTree', auto_param_selection=True, max_iter=10, verbose=False,)order_cells(HSMM_myo)def gm_state(adata): if adata.obs['State'].nunique() <= 1: return 1 t0 = ( adata.obs[adata.obs['Hours'].astype(float) == 0.0]['State'] .astype(int).value_counts() ) return int(t0.idxmax()) if not t0.empty else int(adata.obs['State'].astype(int).iloc[0])order_cells(HSMM_myo, root_state=gm_state(HSMM_myo))print('States:', sorted(HSMM_myo.obs['State'].astype(int).unique().tolist()))
order_cells 拿到 DDRTree 输出之后做四件事(monocle2py/ordering.py)。
第一,把每个细胞投到主图最近的边。closest_vertex[i] 是细胞 i 在 Z 空间里距离最近的 Y 节点;然后枚举该节点的所有相邻边,用线段投影(internal vertex)或直线投影(tip leaf)取最近,记作 P[:,i]。tip leaf 用直线投影是为了让"超出叶端"的细胞延展出去,避免一律截在端点。
第二,在投影点上重建 cell-level MST。N 个投影点的两两欧氏距离 + R 端约定的 min_dist 偏移作为权重,跑 SciPy 的 Kruskal-style MST(scipy.sparse.csgraph.minimum_spanning_tree)。这棵 N-vertex 树才是后续 pseudotime 的载体 —— 它在 Y-level 主图的"骨架"上加了细胞分辨率。
第三,选根节点 + DFS 给出 Pseudotime 与 State。算法是:取这棵树的 weighted diameter(两次最远点搜索),其中一个端点作为 root;以 root 为起点 DFS,沿路径累加边权得 Pseudotime[node],每当当前节点的父节点的度数 > 2(分支点)时,State 编号 +1(ordering.py:127-158)。这个简单递增逻辑直接把"经过分支点"翻译成"换 segment label",所以同一个 trajectory 段内的所有细胞共享一个 State。
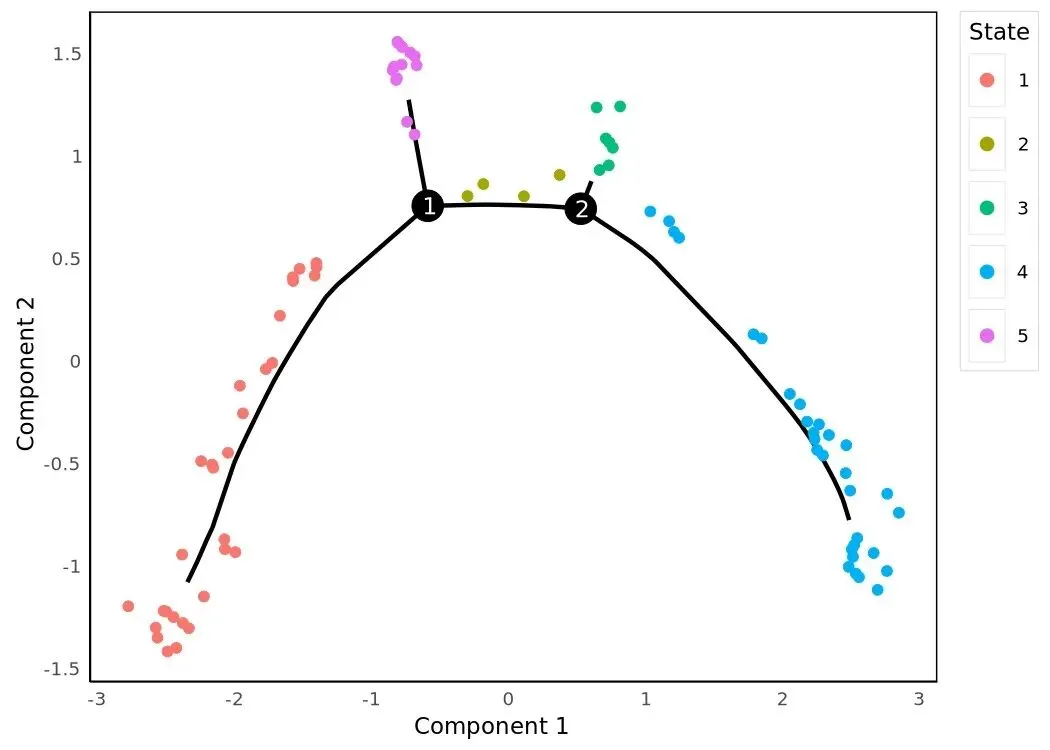
第四,root 重锚定。第一次 order_cells() 调用时根节点是从 diameter 端点里挑的,谁先谁后取决于树的搜索顺序,跟生物学并无直接对应。所以 tutorial 紧接着定义 gm_state(adata):在 Hours==0 的细胞里数哪个 State 最多,把那个 State 当作 root_state 重新调一次 order_cells。这是一种事后用 metadata 校准 trajectory 方向的做法,适合有时间标签的实验。最终得到 5 个 State。
可视化
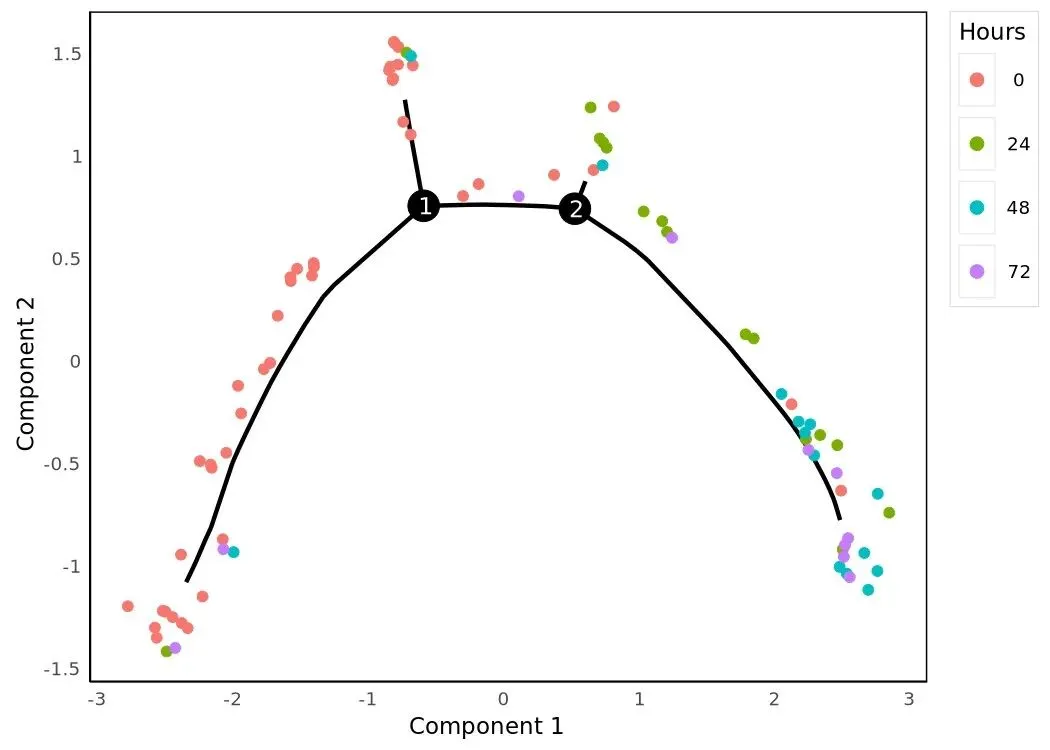
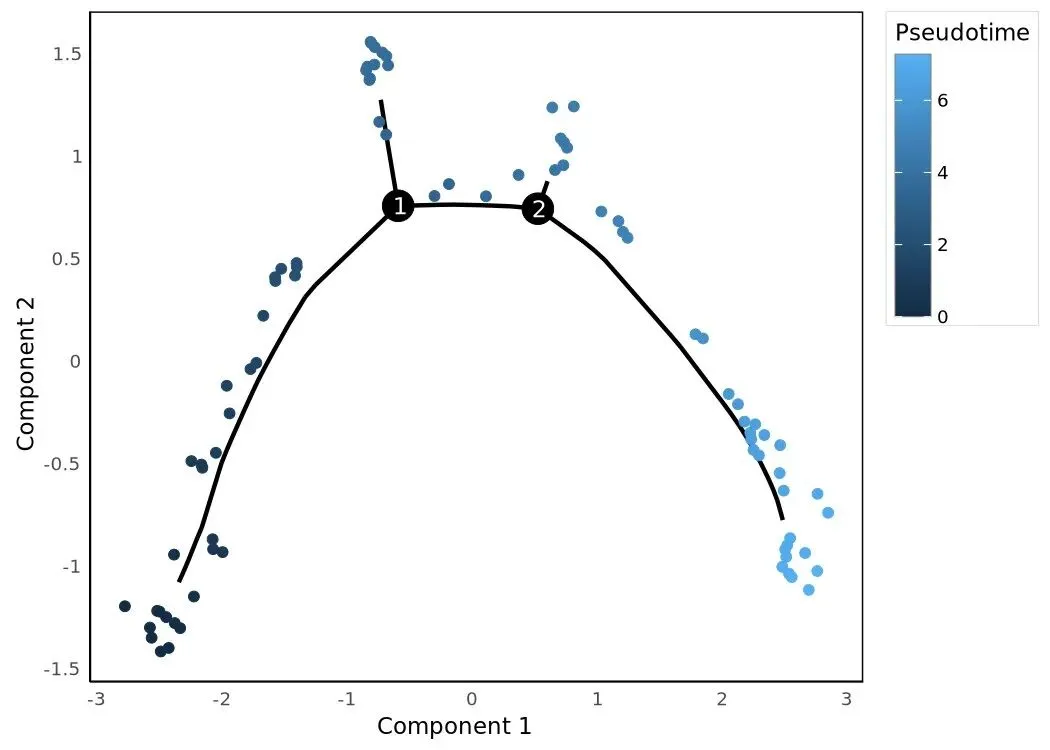
HSMM_myo.obs['Hours'] = HSMM_myo.obs['Hours'].astype('category')plot_cell_trajectory(HSMM_myo, color_by='Hours')
plot_cell_trajectory(HSMM_myo, color_by='State')
plot_cell_trajectory(HSMM_myo, color_by='Pseudotime', show_branch_points=True)
三张 trajectory 是同一棵树、不同上色。Hours 上色直观给出"原始时间点是怎么沿树排布"——理想情况是早时间点聚在 root 端、晚时间点散到 tip。State 上色显示分支结构(每个 segment 一种颜色)。Pseudotime 上色是连续色阶,叠加 branch points 让分叉位置更明确。三张图的几何骨架完全一致,只是上色变量不同。
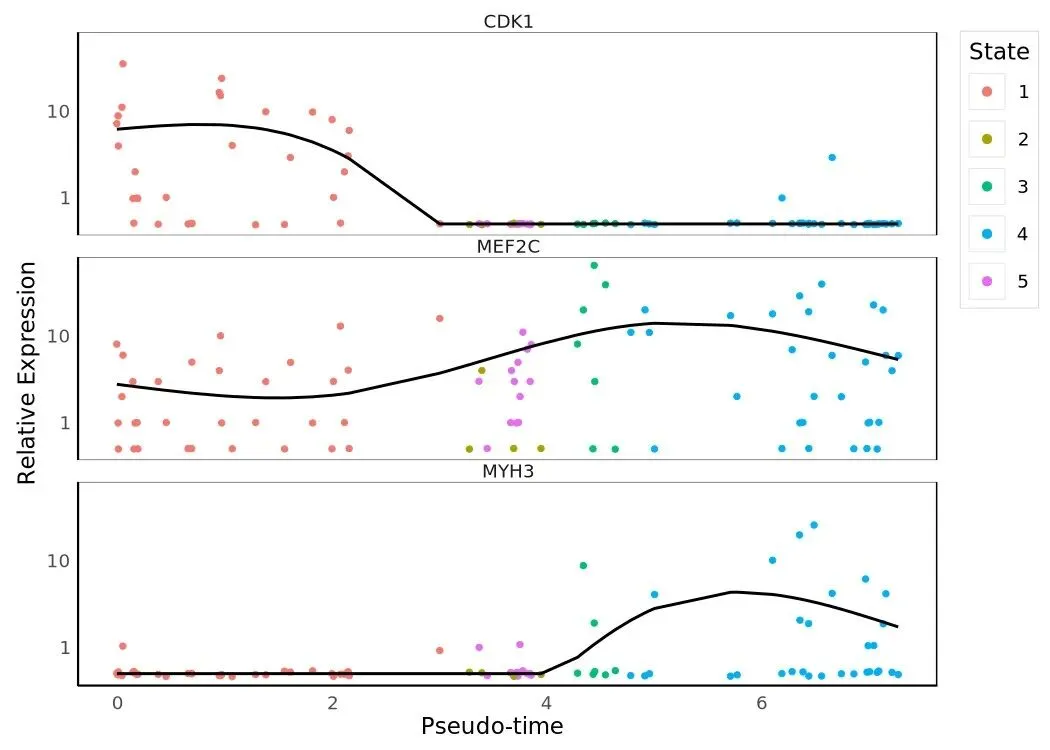
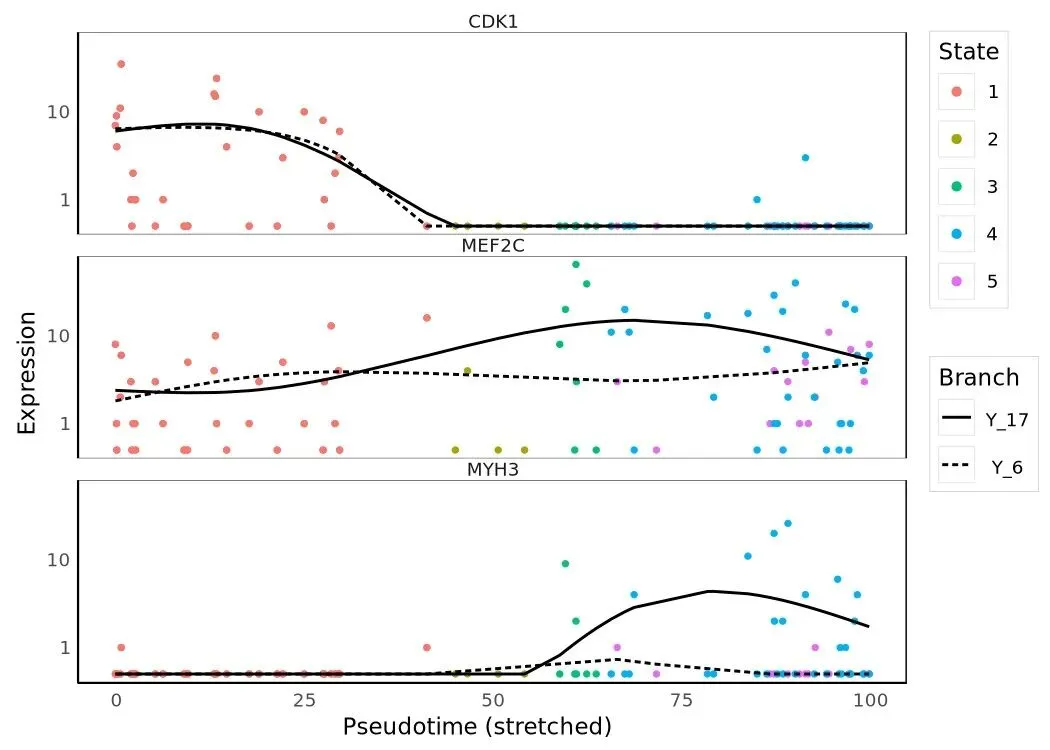
marker_short = ['CDK1', 'MEF2C', 'MYH3']my_genes = HSMM_myo.var_names[ HSMM_myo.var['gene_short_name'].isin(marker_short)].tolist()plot_genes_in_pseudotime(HSMM_myo[:, my_genes])
plot_genes_branched_pseudotime( HSMM_myo[:, my_genes], branch_point=1, ncol=1,)
plot_genes_in_pseudotime 把整条 pseudotime 当作横轴,每个基因画一条 GAM-smoothed 表达曲线;plot_genes_branched_pseudotime 在 branch_point=1 处把 trajectory 一分为二,每条子分支单独画一条曲线、共享 root 段。同样的三个标志基因 CDK1(细胞周期)、MEF2C(早期肌生成转录因子)、MYH3(晚期肌纤维收缩蛋白)在两种画法下能给出不同的解读视角:线性图看整体趋势,branched 图看哪个分支才是"成熟肌纤维"那一支。
Pseudotime heatmap
marker_panel_short = [ 'MEF2C', 'MEF2D', 'MYF5', 'ANPEP', 'PDGFRA', 'MYOG', 'TPM1', 'TPM2', 'MYH2', 'MYH3', 'NCAM1', 'TNNT1', 'TNNT2', 'TNNC1', 'CDK1', 'CDK2', 'CCNB1', 'CCNB2', 'CCND1', 'CCNA1', 'ID1',]marker_genes = HSMM_myo.var_names[ HSMM_myo.var['gene_short_name'].isin(marker_panel_short)].tolist()marker_cds = HSMM_myo[:, marker_genes].copy()marker_cds.uns['monocle2'] = dict(HSMM_myo.uns['monocle2'])set_disp_fit_info( marker_cds, {'disp_func': _disp_func, 'coefficients': {'asymptDisp': 0.05, 'extraPois': 1.0}, 'disp_table': pd.DataFrame()}, name='blind',)pt_test = differential_gene_test(marker_cds, '~sm.ns(Pseudotime, df=3)', '~1')sig_pt = pt_test[pt_test['qval'] < 0.1].sort_values('qval').index.tolist()ph = plot_pseudotime_heatmap( HSMM_myo[:, sig_pt], num_clusters=3, norm_method='log', show_rownames=True,)show(ph, width=5, height=6)
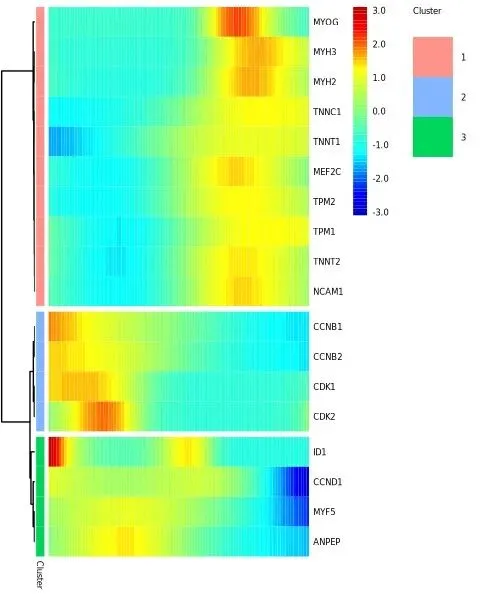
模型公式 ~sm.ns(Pseudotime, df=3) 是自然 cubic spline,3 个内部节点。NB GLM 下的 LRT 比较 full(spline-of-pseudotime + offset)与 reduced(只有 intercept + offset),qval<0.1 的基因被认为表达随 pseudotime 显著变化。用 spline 而不是线性是因为成肌过程的关键基因(如 MYH3)往往呈延迟激增,线性 GLM 拟合不出这种形状。Heatmap 把这些基因按 pseudotime 聚成 3 组,从 root 端的 CDK1/CCNB1 这类周期基因,到中段的 MYF5/MEF2C,再到 tip 端的 MYH3/TNNT1,清楚地呈现了从增殖型 myoblast 到收缩型 myocyte 的程式化转变。
BEAM:分支依赖基因
panel.uns['monocle2'] = dict(HSMM_myo.uns['monocle2'])set_disp_fit_info( panel, {'disp_func': _disp_func, 'coefficients': {'asymptDisp': 0.05, 'extraPois': 1.0}, 'disp_table': pd.DataFrame()}, name='blind',)beam_res = beam(panel, branch_point=1)beam_res = beam_res.sort_values('qval')print('genes tested:', len(beam_res), '| qval<1e-3:', int((beam_res['qval'] < 1e-3).sum()))beam_res[['gene_short_name', 'pval', 'qval']].head(15)
BEAM(Branched Expression Analysis Modeling)在 Y-node(主图质心)层面遍历 trajectory,把每个 Y 节点上挂载的细胞按所属分支(branch A / B)分组,然后做一对模型对比的 LRT:
- full:
~sm.ns(Pseudotime, df=3) * Branch —— pseudotime 与分支标签的交互 - reduced:
~sm.ns(Pseudotime, df=3) —— 不区分分支
LRT 显著的基因就是"在两条分支上随 pseudotime 表达动力学不同"的基因。原理上 BEAM 就是把 trajectory 上的差异表达检验从一维(纯 pseudotime)推广到二维(pseudotime × branch)。HSMM 的 200 基因 panel 在 qval<1e-3 下只命中 2 个 hit(DCN、SLC25A5),数量太少,因此 tutorial 里 plot_genes_branched_heatmap 那个 cell 走 print('Fewer than 5 BEAM hits') 分支没出图 —— 这是细胞数与基因 panel 都偏小时检出力受限的现实,跑全转录组的 ordering filter 通常能给出更丰满的 BEAM 列表。
收尾
到这一步,HSMM tutorial 走完。整条 pipeline 的关键判断都落在两个算法上:DDRTree 学嵌入与树、order_cells 的几何投影 + DFS 给 pseudotime 与 state。差异表达检验(NB GLM + spline + LRT)只是后处理,真正的 trajectory 信息在 Z 与 Y 里。
Bio-Babel/Monocle2-python 的 tutorials 目录还有另外三个 notebook:V2_2_lung_BEAM(BEAM 专项,有充足的分支基因可视化)、V2_3_Olsson_corrected(WT + KO 多条件髓系祖细胞)、V2_4_Paul(造血)。三者的 pipeline 与 V2_1 大同小异,但每一个会突出不同的处理细节(批次效应、condition design、root 选择)。算法层面想再深入的话,推荐去 DDRTree-python 仓库的 tests/test_r_parity_fixes.py、tests/test_backend_torch_gold.py 看怎么用 R 端 fixture 验证 numpy 和 torch 两个 backend 的数值 parity —— 一份足够清晰的 reference test 比 README 更能讲清算法的边界条件。
代码: https://github.com/Bio-Babel/Monocle2-python