肿瘤单细胞代谢预测实战|全流程python分析
- 2026-07-02 22:24:56
编者按
在重构R算法这个栏目中,我们将介绍omicverse开源重构生态最新的成果以及完整的教程,欢迎关注。
本第三期教程里,由首都医科大学的林子恒给大家带来单细胞代谢分析:这是omicverse中
Metabolism模块的内容。我们重构了scmetabolism和scFEA。单细胞转录组测的是RNA,不是代谢物。但很多时候,代谢状态会在转录层面留下非常强的信号。比如糖酵解、氧化磷酸化、戊糖磷酸途径这些通路,如果一类细胞真的在代谢层面被激活,那么相关基因的表达也会跟着发生系统性变化。
本文将复现Xiao, Dai & Locasale 2019在Nature Communications发表的肿瘤微环境单细胞代谢图谱分析。
本文AI率20%,阅读大约需要5min。
OmicVerse团队
1. 引言
肿瘤不是一团均匀的细胞。
一个真实的肿瘤组织里,有恶性细胞、成纤维细胞、内皮细胞、T细胞、B细胞、巨噬细胞、树突细胞。它们待在同一个微环境里,但代谢方式完全不一样。
如果你用bulk RNA-seq去看,得到的是所有细胞混在一起的平均值。这个平均值当然有用,但它会把很多真正有意思的东西抹平。
比如我们真正想问的是:
哪些细胞的糖酵解最强? 恶性细胞是不是整体代谢更活跃? 氧化磷酸化是不是只在某一群肿瘤细胞里升高? 缺氧和糖酵解是不是在单细胞层面相关? 不同免疫/基质细胞是否有各自的代谢偏好?
这些问题都需要单细胞分辨率。
2019年,Xiao, Dai和Locasale在Nature Communications发表了文章,系统分析了肿瘤微环境的单细胞代谢图谱。他们的一个主要结论是:在头颈鳞癌数据中,恶性细胞上调的代谢通路数量最多,肿瘤内部还存在明显的代谢异质性。
2. Metabolism模块能做什么
OmicVerse里统一通过这个接口做代谢分析:
ov.single.Metabolism(adata, method='scmetabolism')
目前这个模块里有三类思路:
scmetabolism | ||
scfea | ||
compass |
在本期教程,我们主要使用 scmetabolism 来计算85条KEGG代谢通路活性,最后再用 scFEA 做一个代谢通量的补充。
scmetabolism是复现的R包,我们在确保计算得分一致性的前提下,对计算速度也进行了较大的优化,最终得到的是一个“细胞 × 代谢通路”的矩阵,我们可以用这个矩阵进行差异分析、UMAP可视化、热图和细胞类型比较等内容。
3. 数据读入
首先载入OmicVerse并设置绘图风格:
import omicverse as ov
ov.plot_set()
这里使用的是Puram et al. 2017的头颈部鳞状细胞癌数据,也就是GSE103322。这个数据包含19位患者的5,578个单细胞,也是Xiao 2019那篇文章分析的数据。
adata = ov.datasets.metabolism_hnsc()
adata
AnnData object with n_obs × n_vars = 5578 × 23686
obs: 'patient', 'malignant', 'celltype', 'lymph_node', 'maxima_enzyme'
uns: 'dataset', 'expression_units'
我们看一下细胞类型组成:
adata.obs['celltype'].value_counts()
celltype
Malignant 2215
Fibroblast 1440
T cell 1237
Endothelial 260
B cell 138
Mast 120
Macrophage 98
Dendritic 51
myocyte 19
Name: count, dtype: int64
在这个数据中,恶性细胞数量足够多,免疫和基质细胞也比较完整,可以直接比较肿瘤细胞和微环境细胞之间的代谢差异。
4. 构建UMAP
由于这个数据已经是标准化后的表达矩阵,所以我们直接选高变基因,然后做scale、PCA、邻居图和UMAP标准的单细胞分析流程。
ov.pp.highly_variable_genes(adata, n_top_genes=2000)
adata.raw = adata
hvg = adata[:, adata.var.highly_variable].copy()
ov.pp.scale(hvg)
ov.pp.pca(hvg, layer='scaled', n_pcs=30)
ov.pp.neighbors(
hvg,
n_neighbors=15,
n_pcs=30,
use_rep='scaled|original|X_pca',
)
ov.pp.umap(hvg)
adata.obsm['X_umap'] = hvg.obsm['X_umap']
首先看一下UMAP上细胞类型和恶性状态的分布:
ov.pl.embedding(
adata,
basis='X_umap',
color=['celltype', 'malignant'],
frameon='small',
wspace=0.5,
)
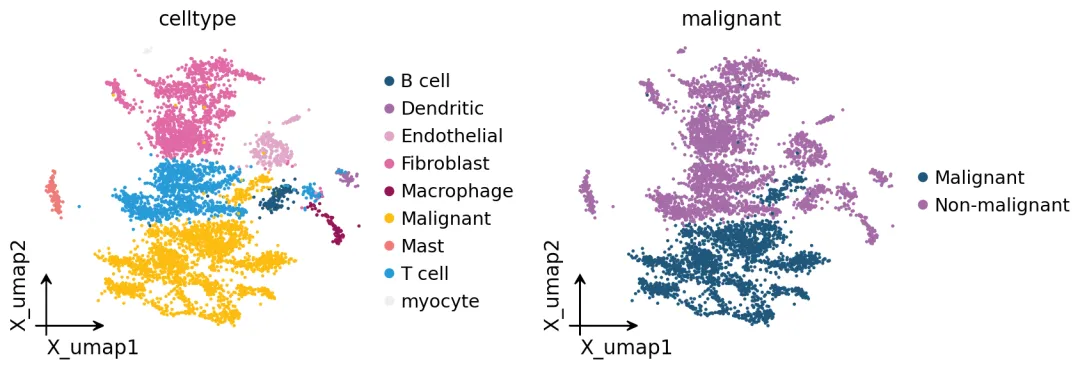
可以看到恶性细胞和其他细胞在UMAP上基本可以分开。后面我们把代谢通路分数投到这个UMAP上,就能看出哪些区域的代谢活性更强。
5. 计算代谢通路活性
接下来运行 scmetabolism。
met = ov.single.Metabolism(adata, method='scmetabolism')
met.run(score_method='AUCell', metabolism_type='KEGG')
这里用AUCell计算所有KEGG代谢通路在每个细胞里的活性。结果会写入:
adata.obsm['X_metabolism']
看一下矩阵大小:
adata.obsm['X_metabolism'].shape
(5578, 85)
也就是说,我们现在得到了一个5,578个细胞 × 85条KEGG代谢通路的活性矩阵。
这一步其实就是后面所有代谢分析的基础。
6. 恶性细胞上调最多代谢通路
Xiao 2019的一个主要结论是:恶性细胞上调的代谢通路数量最多。
我们可以用 ov.single.differential_metabolism 比较恶性细胞和非恶性细胞的代谢通路活性,这个算法调用的也是omicverse重构生态的成果py-limma包。
deg = ov.single.differential_metabolism(
adata,
groupby='malignant',
group1='Malignant',
)
up = deg.query('padj < 0.05 and log2fc > 0')
print(f'{len(up)} of {len(deg)} KEGG pathways up-regulated in malignant cells')
53 of 85 KEGG pathways up-regulated in malignant cells
在这个数据中,我们发现在85条KEGG代谢通路里,有53条在恶性细胞中显著上调。
我们看一下排名靠前的通路:
deg.head(8)
我觉得这个结果还是比较符合肿瘤代谢的背景知识。糖酵解、戊糖磷酸途径、氧化磷酸化、嘧啶代谢、嘌呤代谢都排在前面。
接着我们把不同细胞类型分别拿出来比较,统计每类细胞显著上调的代谢通路数量:
import pandas as pd
n_up = {
ct: len(
ov.single.differential_metabolism(
adata,
groupby='celltype',
group1=ct,
).query('padj < 0.05 and log2fc > 0')
)
for ct in ['Malignant', 'Fibroblast', 'Macrophage', 'B cell', 'T cell']
}
pd.Series(n_up).sort_values(ascending=False)
Malignant 53
Macrophage 38
Fibroblast 27
B cell 17
T cell 15
dtype: int64
可视化:
import matplotlib.pyplot as plt
ax = pd.Series(n_up).sort_values().plot.barh(
color='#C44E52',
figsize=(5, 3),
)
ax.set_xlabel('# KEGG pathways up-regulated')
ax.set_title('Metabolic activity across the microenvironment')
plt.show()
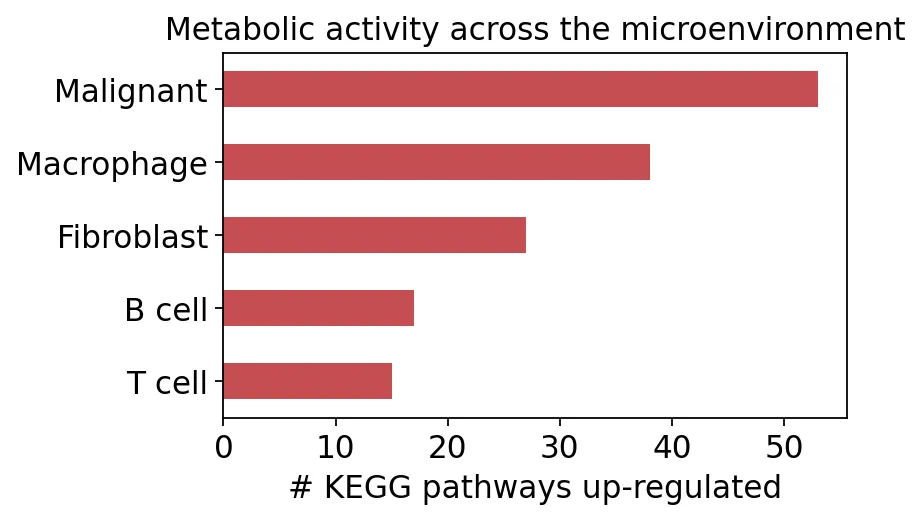
我们发现,恶性细胞上调通路数量最多,巨噬细胞其次,T细胞和B细胞相对更少。
7. 肿瘤内部的氧化磷酸化异质性
代谢分析里,我们除了分析出“恶性细胞代谢更活跃”这个结论外,我们还可以进一步去分析代谢异质性。例如,同样是恶性细胞,代谢状态也不一样。
这里我们把氧化磷酸化通路提取到 adata.obs 中:
met.to_obs('Oxidative phosphorylation')
ov.pl.embedding(
adata,
basis='X_umap',
color='Oxidative phosphorylation',
cmap='Reds',
frameon='small',
)
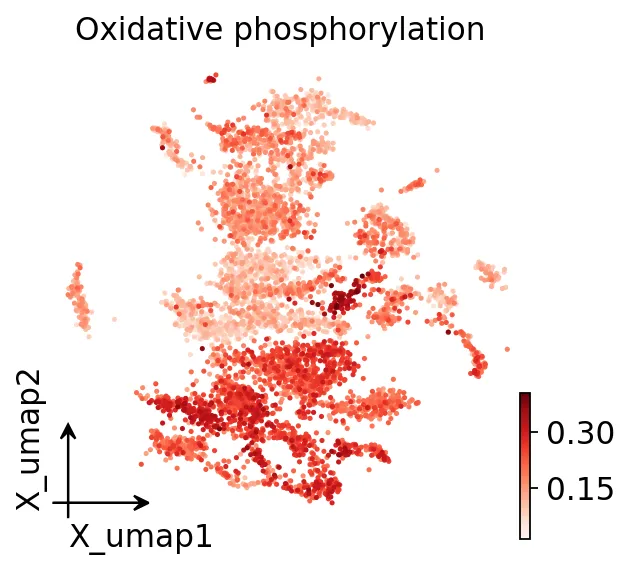
从UMAP上可以看到,氧化磷酸化没有在所有恶性细胞里均匀升高,只有部分区域更强。
再按细胞类型画小提琴图:
ov.pl.violin(
adata,
keys='Oxidative phosphorylation',
groupby='celltype',
rotation=90,
)
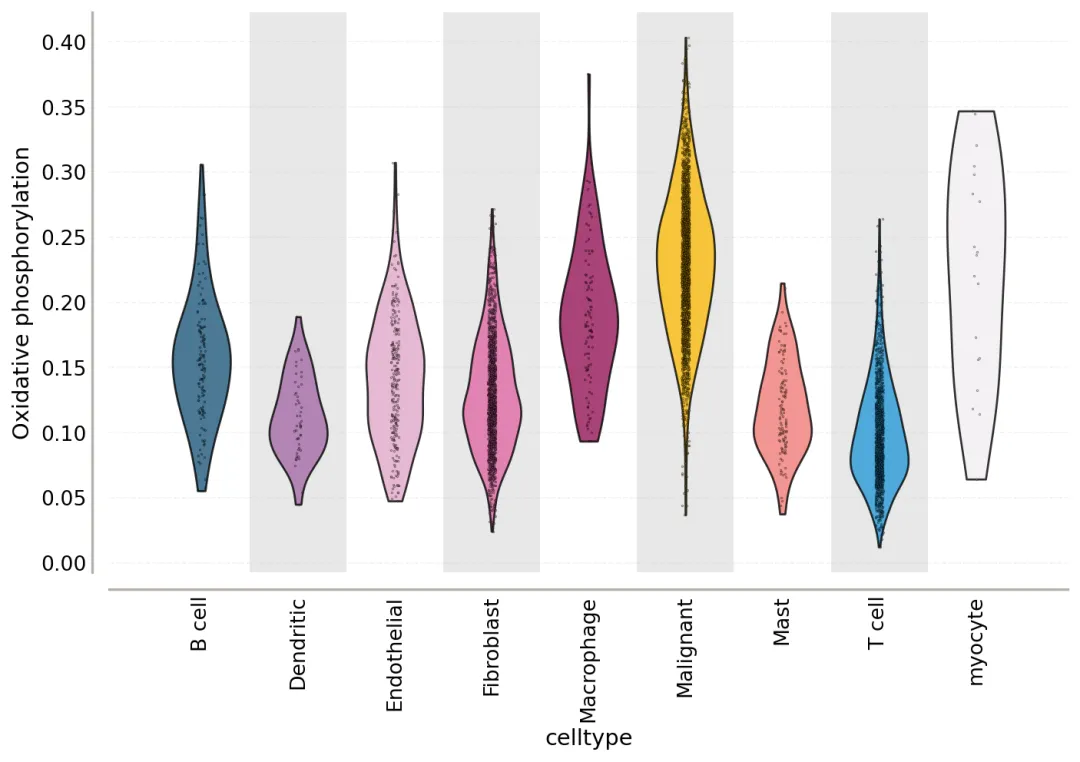
我们从这张图可以获得两个层次的信息。
第一,不同细胞类型之间的氧化磷酸化水平不一样。 第二,恶性细胞内部的分布也很不均匀。这就是我们常说的肿瘤内部代谢异质性。也就是说,肿瘤细胞不是一整块共同切换到同一种代谢模式,而是内部本身就有不同代谢状态的细胞。
8. 缺氧、糖酵解和氧化磷酸化
除此之外,提到肿瘤代谢,很多人会想到Warburg效应:肿瘤细胞即使在有氧条件下,也倾向于使用糖酵解。
于是一个很自然的问题是,糖酵解和氧化磷酸化在单细胞层面到底是什么关系?
我们首先用HALLMARK HYPOXIA基因集给每个细胞计算缺氧分数,这里用到了上期教程里的ov.es.aucell函数:
sigs = ov.utils.geneset_prepare(
ov.utils.predefined_signatures['hallmark'],
organism='Human',
)
ov.es.aucell(
adata,
signatures={'hypoxia': sigs['HALLMARK_HYPOXIA']},
)
计算完成后,我们从中提取缺氧、糖酵解和氧化磷酸化三个分数:
scores = met.get()
energy = pd.DataFrame({
'hypoxia': adata.obsm['score_aucell']['hypoxia'].to_numpy(),
'Glycolysis': scores['Glycolysis / Gluconeogenesis'].to_numpy(),
'OXPHOS': scores['Oxidative phosphorylation'].to_numpy(),
})
energy.corr().round(2)
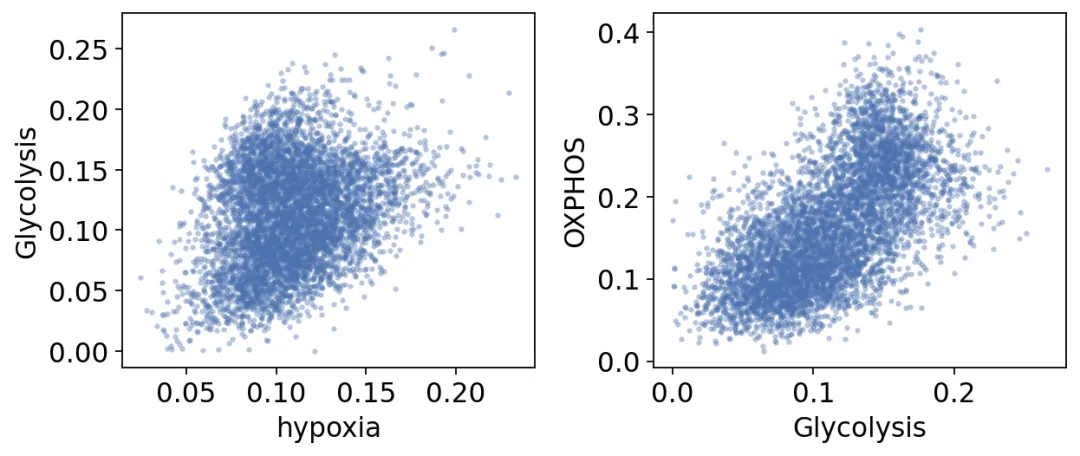
hypoxia Glycolysis OXPHOS
hypoxia 1.00 0.29 -0.02
Glycolysis 0.29 1.00 0.62
OXPHOS -0.02 0.62 1.00
我们发现,
第一,缺氧和糖酵解是正相关的,相关系数为0.29。这符合HIF轴激活糖酵解的经典认知。 第二,糖酵解和氧化磷酸化也是正相关的,相关系数为0.62。也就是说,在这个数据中,两者不是简单的一开一关关系,也可以同时升高。
我们画一个散点图来看一下:
fig, axes = plt.subplots(1, 2, figsize=(8, 3.5))
for ax, (x, y) in zip(
axes,
[('hypoxia', 'Glycolysis'), ('Glycolysis', 'OXPHOS')],
):
ax.scatter(energy[x], energy[y], s=4, alpha=0.3, color='#4C72B0')
ax.set(xlabel=x, ylabel=y)
plt.tight_layout()
plt.show()
9. 不同细胞类型的代谢程序
除了恶性细胞,微环境里的不同细胞类型也有自己的代谢偏好。
这里使用 ov.pl.metabolism_heatmap,对每个细胞类型内的通路活性做平均,然后筛选区分度较高的代谢通路画热图。
ov.pl.metabolism_heatmap(
adata,
groupby='celltype',
n_features=25,
)
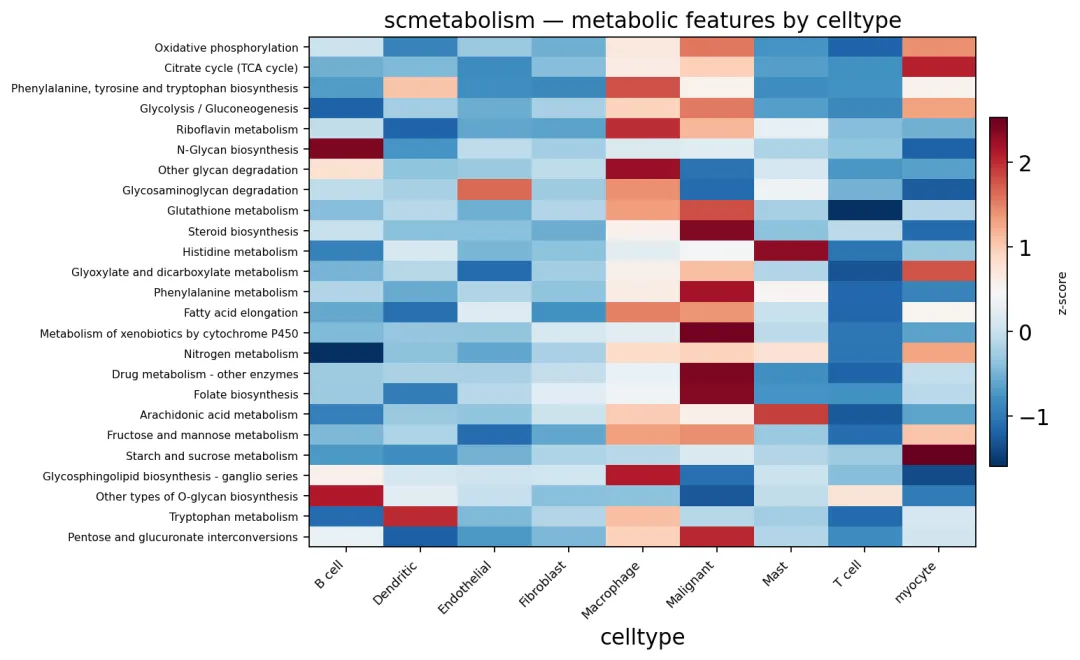
一般来说,这张图适合放在结果部分当总览图。
如果你在自己的项目里做肿瘤微环境分析,我建议可以画一张类似的热图。因为它可以快速告诉读者:哪些代谢通路是恶性细胞偏高,哪些是免疫细胞偏高,哪些是成纤维细胞偏高。
10. 用scFEA估计代谢通量
最后,前面的 scmetabolism 计算的是通路活性,其实还是基因集打分。
但代谢分析里还有另一个概念:通量。
对于通量,它想回答的是,一个代谢反应或者代谢模块到底跑得有多快。scFEA就是这类方法,它用图神经网络和代谢物质量平衡约束,估计大约168个代谢模块的通量。
由于scFEA计算比通路打分更重,因此在这里随机抽取400个细胞做演示:
import numpy as np
idx = np.random.default_rng(0).choice(adata.n_obs, 400, replace=False)
sub = adata[idx].copy()
flux = ov.single.Metabolism(sub, method='scfea')
flux.run(n_epoch=25, verbose=False)
我们也可以画相似的代谢特征热图:
ov.pl.metabolism_heatmap(
sub,
groupby='celltype',
n_features=20,
)
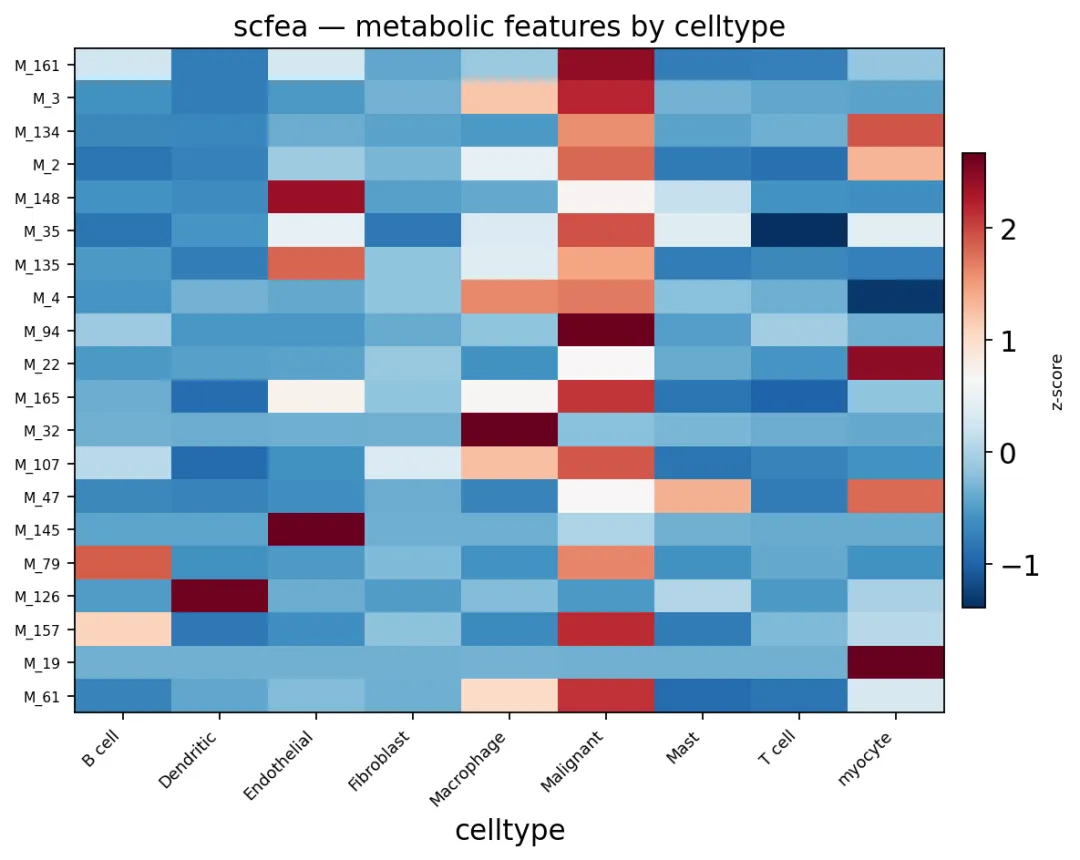
总的来说,scFEA和scMetabolism看的角度不一样。前者更接近代谢模块通量,后者更接近通路活性。
11. 总结
在本期教程中,我们用Puram 2017头颈鳞癌单细胞数据,复现了Xiao 2019单细胞代谢图谱中的几个主要结论:
使用 ov.single.Metabolism(method='scmetabolism')计算了5,578个细胞的85条KEGG代谢通路活性。恶性细胞中有53条KEGG代谢通路显著上调,是所有细胞类型里最多的。 氧化磷酸化在肿瘤细胞内部并不均匀,提示恶性细胞内部存在代谢异质性。 缺氧和糖酵解正相关,糖酵解和氧化磷酸化也正相关,说明两种能量代谢程序可以共同升高。 使用 scFEA可以进一步从通路活性走向代谢通量分析。
实际上我觉得代谢分析,会是单细胞肿瘤研究里越来越常见的一部分。因为只做细胞类型注释已经不太够了,我们还需要知道这些细胞到底处在什么功能状态里。在实际应用中,空转数据也可以一键应用我们的算法预测代谢情况。
代谢状态就是功能状态里绕不开的一类。
12. 交流群
最后,如果你也很好奇我们omicverse生态的最新进展,想第一时间体验到新功能,欢迎加入我们的交流群~

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- VASP新手入门,对于VASP以及Linux系统初学者的福音~(附VASP简单结构优化的详细过程)
- 不会用ps处理图像?没关系,我可以用python,一样可以稳稳拿下!
- 我好棒呀,自己安装了Python解释器!
- TextBlob,一个超好用的Python文本分析库!
- 5个珍藏极品Python网站,请偷偷实用!
- 5种Python爬虫用法,你知道几种?
- 暑假弯道超车双王牌!15天玩转Python人工智能+无人机,思维能力+专注力双提升
- 跳出Python��拥抱世界
- 摩根士丹利 | Python全栈开发工程师 - Associate - 网络安全工程
- 14个Python自动化脚本,工作效率提升100%
