本文全文约 3800 字,预计阅读 12 分钟
解读文献基础信息 文献标题:MSIDAT: an automated platform for improved metabolite annotation in mass spectrometry imaging via mass shift evaluation and customized databases 期刊:Analytical and Bioanalytical Chemistry(Springer) DOI:https://doi.org/10.1007/s00216-026-06576-1通讯单位:北京协和医学院北京协和医院检验科、复杂重症罕见病国家重点实验室 核心作者:朱颖、邱玲、于松林;工具开源地址:https://github.com/Yingzhu96/MSIDAT |
一、前言:空间代谢组 MSI 当下最头疼的两大分析难题
质谱成像(MSI / 空间分辨代谢组)可以直接在组织切片原位定位代谢物分布,是肿瘤异质性、临床标志物挖掘的核心手段。但一线做实验、做数据分析的同学一定都踩过两大坑:
1.通用公共数据库匹配假阳性泛滥HMDB、LIPID MAPS 库化合物包罗万象,但没有组织特异性,匹配时会一次性抛出几十种候选代谢物;现有工具要么用固定内置库、无法适配肿瘤 / 器官特殊样本,缺少自建样本专属数据库功能。
2.m/z 质量偏移无法统一适配不同批次、不同 AFADESI/MALDI 仪器会产生稳定的质谱漂移,固定 ppm 容忍度要么大量真实峰漏检,要么杂峰疯狂匹配,手动调参重复性极差。
既往主流工具 METASPACE、MSIannotator、rMSIannotation 只能解决部分问题,无法同时完成「自建数据库 + 数据专属偏移校正」。 本研究基于 Python 开发全自动化开源工具 MSIDAT,一套流程走完数据库构建、偏移评估、智能注释,并用直肠癌临床队列完整验证工具实用性。
二、核心工具三大模块:一站式 MSI 数据处理流水线
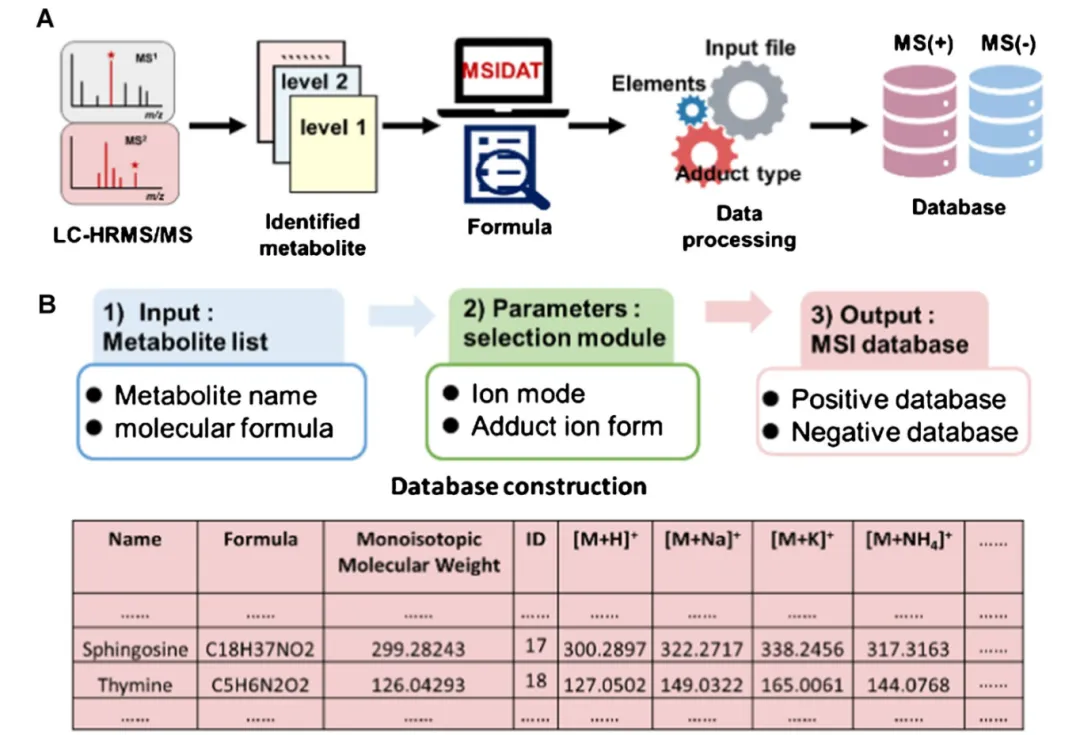
2.1 模块 1:样本专属定制化数据库构建
传统库的缺陷是 “泛而不准”,MSIDAT 直接依托 LC-MS/MS 鉴定的本组织真实代谢物生成专属离子库。
操作逻辑极简
1.输入:仅 Excel 两列 —— 代谢名称 + 分子式(数据来自样本 LC-MS/MS 二级谱鉴定结果)
2.自动批量计算
○解析分子式,计算单同位素精确分子量
○批量生成正负模式全部加合离子理论 m/z
○正模式 6 类加合:[M+H]⁺、[M+Na]⁺、[M+K]⁺、[M+NH₄]⁺、[M-H₂O+H]⁺、[M]⁺
○负模式 5 类加合:[M-H]⁻、[M-H₂O-H]⁻、[M+Cl]⁻、[M+CH₃COO]⁻、[M+HCOO]⁻
3.输出:分正负模式数据库 Excel,包含 ID、分子量、全部理论 m/z
实测效率
本研究 1419 个直肠癌代谢物,一次性生成 14055 条离子记录,运算耗时不足 5 秒。
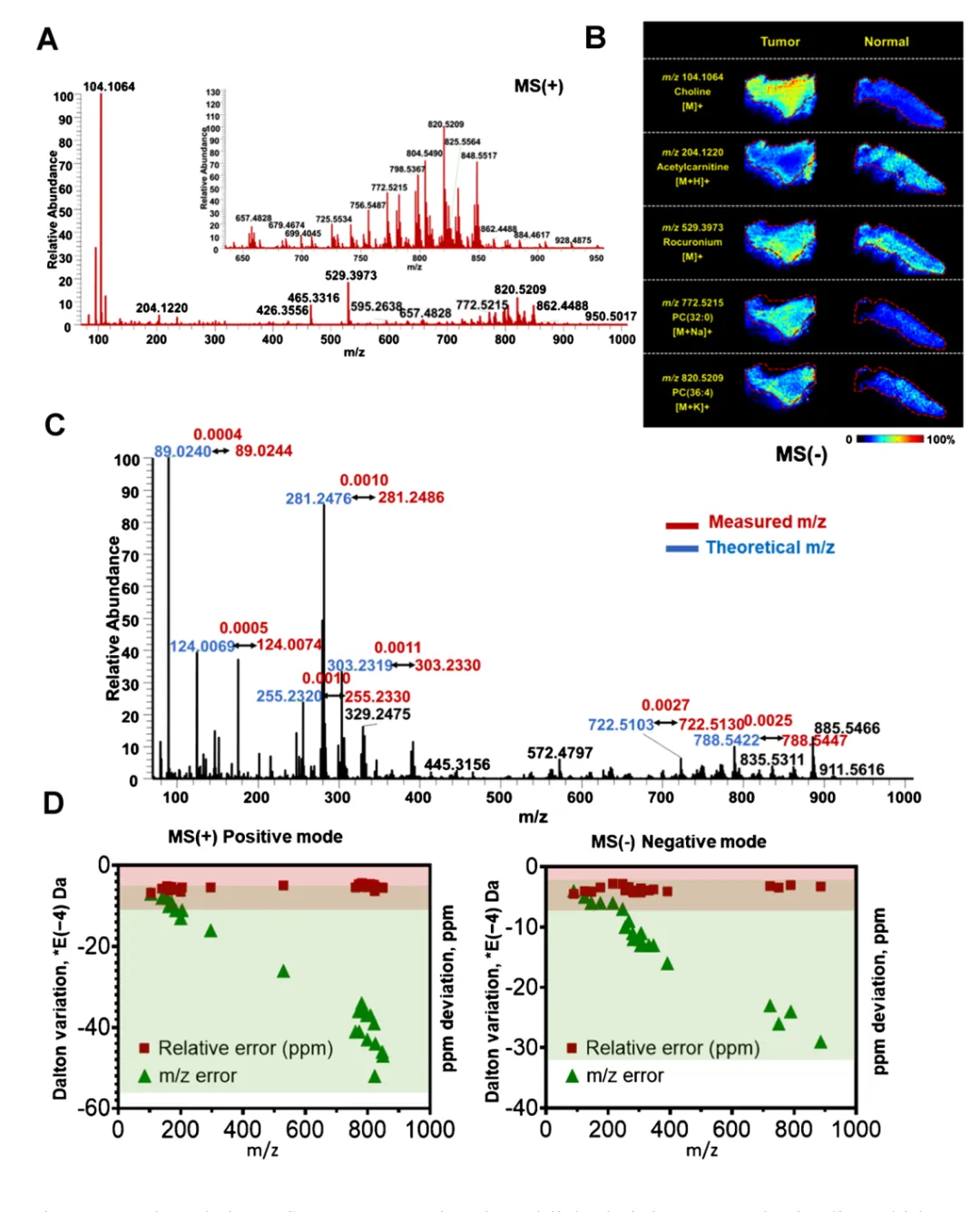
2.2 模块 2:基于 44 个通用内源离子的 m/z 偏移定量评估
这是全文最核心创新点:不修改原始质谱轴,仅用内源参考离子量化整套数据漂移区间,自适应调整匹配窗口。
44 个参考离子完整筛选标准(硬核技术细节)
很多读者会好奇这 44 个通用离子怎么筛选、怎么保证无假峰,筛选 6 层质控标准如下:
1.ROI 扣背景:仅保留组织特有离子,剔除溶剂、仪器外源噪音;
2.强度过滤:平均离子强度>1000,仅高信噪比主峰;
3.代谢物定性:仅保留有明确分子式的内源代谢物,排除未知杂峰;
4.空间成像验证:离子信号完整贴合组织轮廓、全片广泛分布;
5.全质量覆盖:70–1000 Da 均匀分配,正模式 23 条、负模式 21 条,合计 44 条;
6.跨组织验证:在小鼠脑 / 肝 / 肾 / 心 / 肺等 8 种器官 + 血清全部稳定检出,通用性拉满。
偏移计算核心规律
1.绝对误差(Da)随 m/z 升高持续变大;
2.相对误差(ppm)全质量区间高度稳定; 本直肠癌数据集实测偏移:正模式 - 6.72 ~ -4.36 ppm,负模式 - 5 ~ -2 ppm。 基于该区间放宽匹配容忍度,兼顾注释覆盖度与假阳性控制。
补充:44 条离子完整清单未放在正文,全部收录于文章补充材料 Table S3,GitHub 工具包附带标准参考离子 Excel,可直接复用。 |
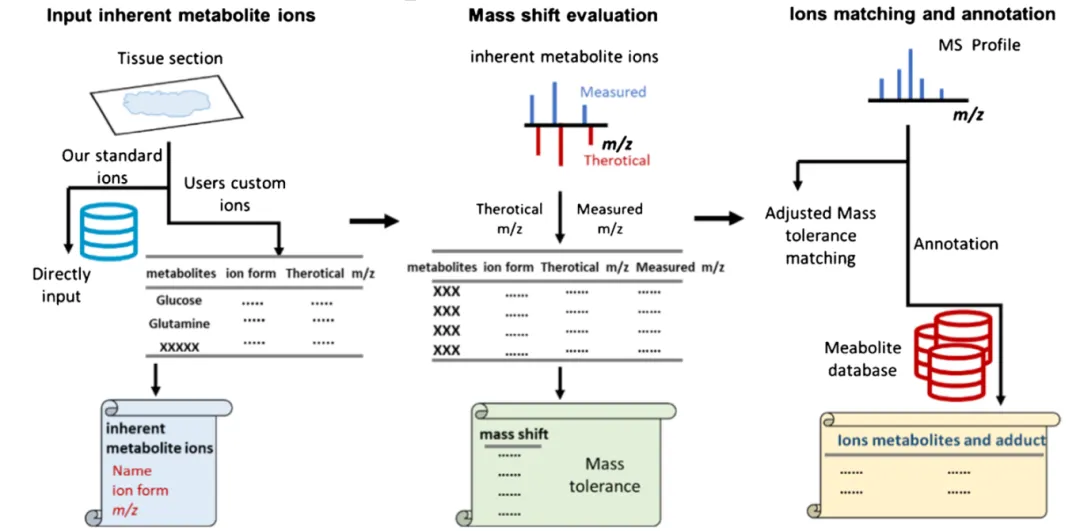
2.3 模块 3:质量误差驱动智能代谢物注释
偏移评估得到 ppm 区间后,导入定制数据库自动批量匹配:
1.输入文件:MSI 全谱峰表 + 自建样本数据库;
2.参数设置:根据偏移结果设置专属 ppm 容忍度,摒弃固定窗口;
3.输出完整注释表:实测 m/z、匹配代谢物、加合形式、理论 m/z、匹配误差;
工具对比优势
相比 MSIannotator 等传统软件,MSIDAT 三合一集成: ✅ 可自建样本专属库(降低假阳性) ✅ 内源离子自动评估数据专属漂移 ✅ 全流程自动化,无代码门槛,纯 Excel 交互 ❌ 现有工具仅支持公共库 / 固定库,无偏移自适应校正
三、实验体系:直肠癌临床队列完整验证工具性能
3.1 临床样本与仪器平台
1.临床样本
○11 例直肠癌患者 21 份组织:AFADESI-MSI 空间成像;
○2 例患者 4 份组织:LC-MS/MS 非靶向代谢组(用于建库); 伦理批号 I-25PJ0484,入组标准:原发直肠腺癌、无远处转移、术前未化疗。
2.仪器配置
○LC-MS/MS:Vanquish UHPLC + Orbitrap Exploris 120;
○AFADESI-MSI:Orbitrap Exploris 240,空间步长 0.2 mm。
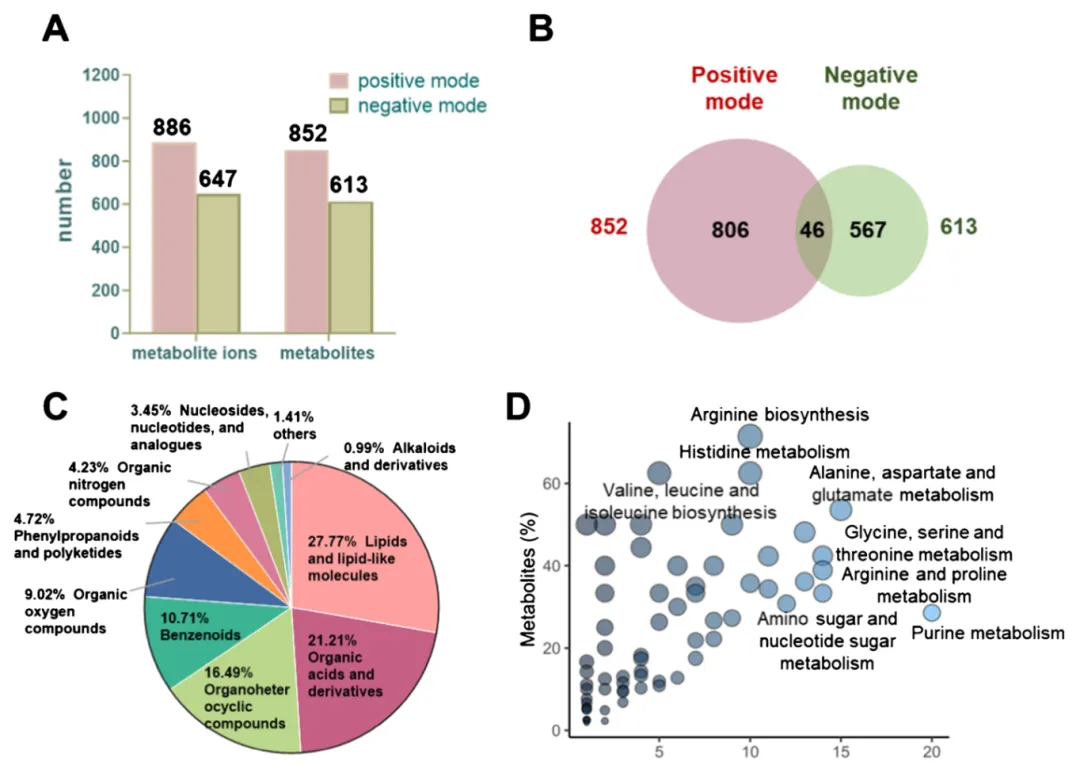
3.2 基础代谢全景:1419 个高置信度代谢物
1.正负模式合计 1419 个 Level1/Level2 高置信代谢物;
2.覆盖脂质、有机酸、核苷、氨基酸等 8 大类,脂质占比最高;
3.共注释 65 条代谢通路,37 条通路包含≥4 个检出代谢物。 这批代谢物是定制数据库的原始素材,从源头规避无关化合物匹配。
3.3 生物学产出:直肠癌脂肪酸代谢空间重编程
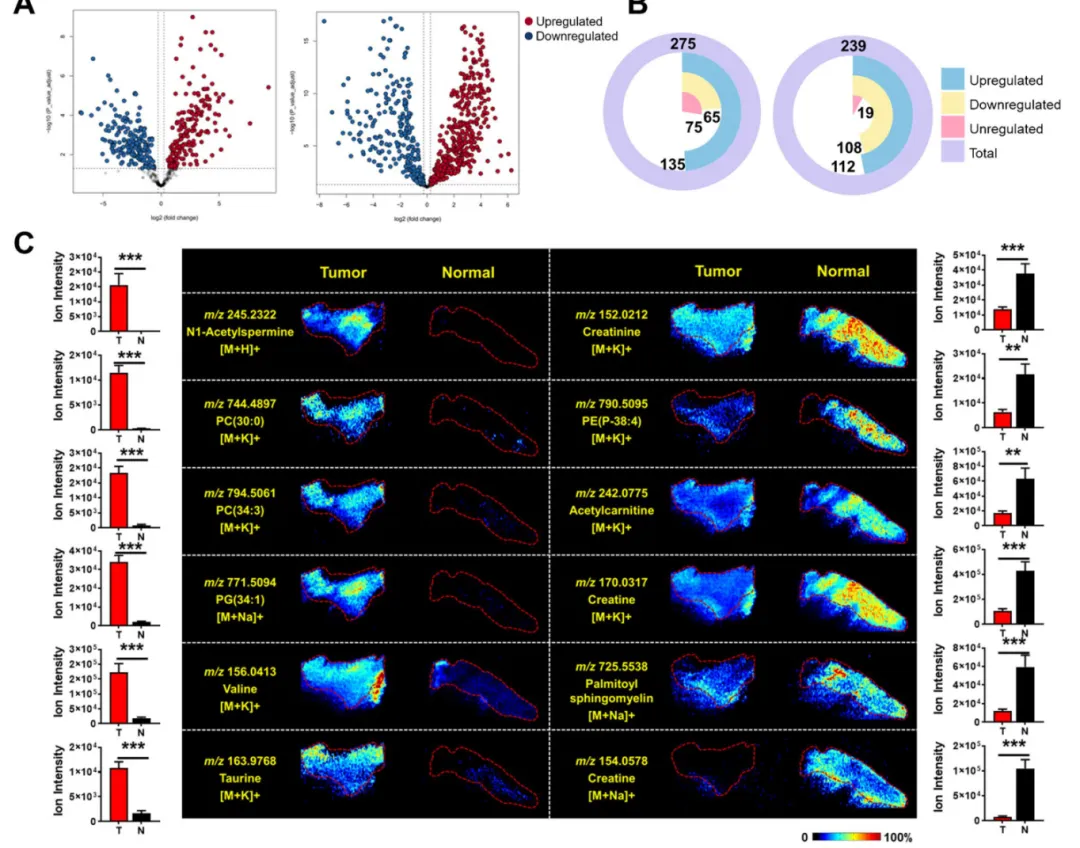
利用 MSIDAT 注释结果做肿瘤 / 癌旁差异分析:
1.正模式 200 个、负模式 220 个离子显著差异(FDR p<0.05);
2.肿瘤上调:多种磷脂、缬氨酸、牛磺酸、多不饱和脂肪酸;
3.肿瘤下调:肌酸、乙酰肉碱、鞘磷脂;
4.核心结论:7 种脂肪酸(油酸、亚油酸、棕榈酸、DHA 等)在肿瘤区域特异性富集,印证结直肠癌脂肪酸合成代谢异常。
四、工具局限与未来拓展方向
现有不足(客观硬核点评)
1.验证以 AFADESI-MSI 数据为主,MALDI-MSI 因基质干扰,内源离子匹配稳定性仍需更多数据集验证;
2.输出仅为推定注释,最终代谢物确认仍需标准品、二级谱、离子淌度正交验证;
后续功能规划
作者团队计划新增:Kendrick 质量缺陷计算、代谢物空间共定位自动化模块,持续更新 GitHub 开源代码。
五、全文总结:MSIDAT 给领域带来的价值
1. 方法学创新
全球首个同时整合「定制代谢库构建 + 内源离子 m/z 漂移自适应校正」的开源 MSI 自动化分析 Python 工具,直击领域两大核心痛点。
2. 使用门槛极低
无需深厚生信 / 编程基础,全部输入输出为 Excel,数秒完成万级离子批量运算,适合临床实验室、高校代谢组课题组日常使用。
3. 临床转化潜力
在直肠癌临床组织中完整实现「建库→偏移校正→注释→空间差异挖掘」完整闭环,为肿瘤空间代谢标志物筛选提供标准化分析流程,可拓展至肺癌、肝癌等各类实体瘤 MSI 研究。
补充资源
1.MSIDAT 开源 GitHub:https://github.com/Yingzhu96/MSIDAT
2.原文补充材料 Table S3:44 个内源参考离子完整清单
3.配套示例数据集、输入模板均随代码开源,可直接上手复现文中直肠癌分析流程

