搞定热图代码!AI助力生信图谱绘制实战
- 2026-07-06 11:16:42
🔥 AI辅助搞定热图代码!AI助力生信图谱绘制实战
还在为复杂的生物信息学图表熬夜写代码?让AI成为你的科研加速器!
你是否也曾面对这样的场景:手头有一批重要的生物信息学数据,需要绘制一张能够清晰展示基因表达模式与临床特征关联的热图,但面对R语言的各种参数和包却感到无从下手?
今天,就让我们一起来看看如何通过自然语言与AI对话,快速生成专业级的热图代码,彻底改变你的科研工作流!
其实,AI辅助绘制热图只是一个简单的实例:在之前稳妥少年公众号分享的很多小工具,包括qvina2/vina 高通量分子对接平台(Shiny):一键完成虚拟药物筛选、稳妥少年——分子动力学模拟结果xvg文件可视化分析平台,一键出图(免费开源)、chatGPT真能助我一臂之力!!!、DeepSeek辅助数据库搭建有多强(R语言Shiny)、请签收!DeepSeek帮您整理了单细胞注释Marker。
都有AI的强力加持!!!
一、科研人的共同痛点:数据可视化之困
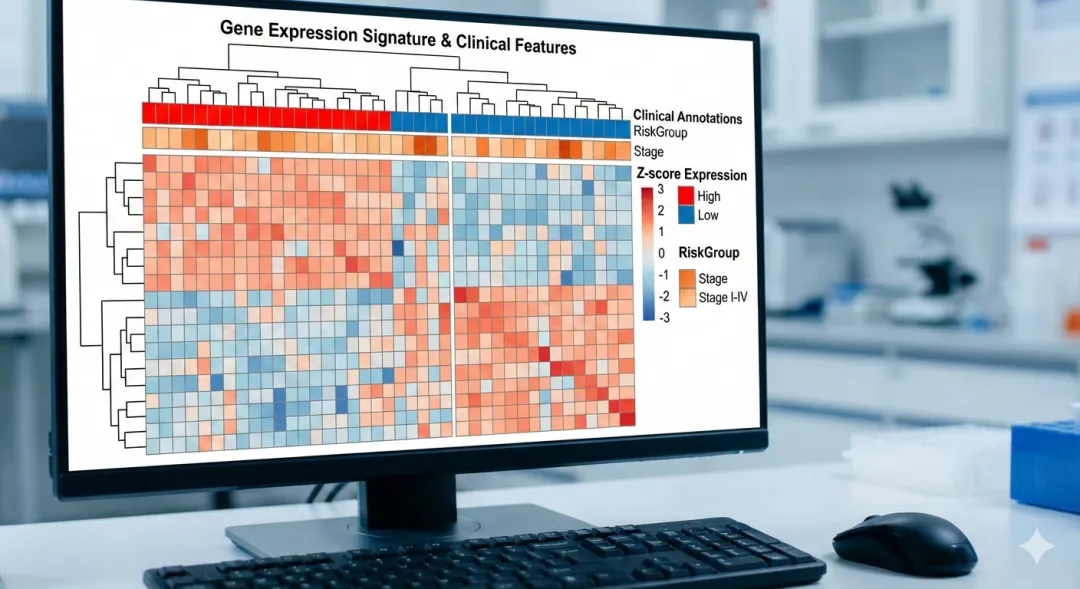
假设你手头有这样一组数据(TCGA),包含了关键基因的表达量、患者的临床信息以及计算出的风险评分。
你需要绘制一张热图:
横坐标:不同的样本分组 纵坐标:基因的表达量 颜色填充:基因表达量高低 分组展示:按照风险等级等临床变量进行颜色标注
传统的做法可能是:打开搜索引擎,查找pheatmap或ComplexHeatmap包的教程,逐一调试参数,处理各种报错……整个过程可能需要数小时甚至更久。
二、AI辅助:用对话代替编码
现在,让我们看看如何通过与AI对话来解决这个问题。
你只需要向AI(如DeepSeek等智能助手)清晰地描述你的需求和数据情况:
你的提示词:
> head(data)
X age fiage or_grade laity OS
1 1331-01A Age >= 60 Stage IIIC G3 Bilateral 1
2 1337-01A Age >= 60 Stage IIIC G2 Bilateral 1
3 1338-01A Age >= 60 Stage IIIC G3 Bilateral 0
4 1343-01A Age >= 60 Stage IV G3 Bilateral 1
5 1347-01A Age >= 60 Stage IV G3 Bilateral 0
6 1350-01A Age < 60 Stage IIIC G3 Bilateral 1
OS.time CACN PD0A SI2 CR1 PTL4 GER2
1 1336 0.36289064 0.13185285 3.751399 3.908957 1.441696 1.84486768
2 61 1.07724300 0.21387604 3.725872 4.267086 2.267566 2.37975957
3 1418 0.36826548 0.23167816 2.467358 4.185978 3.105075 0.95620538
4 361 0.32031137 0.10312832 4.584776 2.837096 3.078985 1.45685871
5 1919 0.17415071 0.03660883 3.362891 1.549226 0.766468 0.43978269
6 1946 0.02133758 0.04404433 2.600294 0.860049 1.893013 0.06295061
AL PR1 TP1 LK G20 T1 Score
1 4.877901 3.744915 3.016978 3.585323 1.2604473 2.768438 -0.4578066
2 3.300739 3.024018 3.886199 1.230633 1.2195867 1.727441 0.1313294
3 5.089651 8.553733 3.210654 2.976419 0.9160944 2.095148 -0.5933797
4 5.241493 2.643856 3.117313 1.762051 2.5082765 1.726744 -0.2480642
5 7.270667 3.541317 1.594740 1.817951 0.7955176 2.133662 -0.7351224
6 6.016113 2.458881 1.692516 2.399171 0.7348288 2.778629 -0.9390338
risk
1 high risk
2 high risk
3 high risk
4 high risk
5 low risk
6 low risk
>
-----
对于这个数据我需要绘制一张热图,横坐标为不同的分组情况,颜色也需要按照不同分组的情况确定,随后纵坐标为12个基因的表达量,颜色填充就是用基因的表达量即可。添加图注,不同分组及不同的变量颜色展示。开始撰写R语言脚本
注:提示词很重要,就是你要描述清楚数据,可以复制一小部分,然后描述清楚你需要绘制的图片内容,就好
AI反馈:
# 加载必要的包
library(pheatmap) # 用于绘制热图
library(RColorBrewer) # 用于颜色调色板
# 设置工作目录(如果需要的话)
# setwd("your/directory/path")
# 1. 数据准备 ----------------------------------------------------------------
# 假设你的数据框名为data
# 提取12个基因的表达数据
gene_names <- c("CA1C", "P0A")
# 提取基因表达矩阵(转置,使基因在行,样本在列)
expression_matrix <- as.matrix(data[, gene_names])
rownames(expression_matrix) <- data$X # 设置行名为样本ID
expression_matrix <- t(expression_matrix) # 转置,使基因在行,样本在列
# 2. 样本分组信息 ------------------------------------------------------------
# 创建分组注释数据框
annotation_col <- data.frame(
Risk_Group = factor(data$risk, levels = c("low risk", "high risk")),
FIGO_Stage = data$stage,
Tumor_Grade = data$rade,
Laterality = data$laity,
Age_Group = data$age,
row.names = data$X
)
# 3. 颜色设置 ----------------------------------------------------------------
# 为风险组设置颜色
risk_colors <- c("low risk" = "blue", "high risk" = "red")
# 为其他临床变量设置颜色(使用Set3调色板,最多12种颜色)
# 阶段颜色
figo_stages <- unique(data$figo_stage)
figo_colors <- brewer.pal(min(length(figo_stages), 9), "Set3")[1:length(figo_stages)]
names(figo_colors) <- figo_stages
#分级颜色
tumor_grades <- unique(data$tumor_grade)
grade_colors <- brewer.pal(min(length(tumor_grades), 9), "Set2")[1:length(tumor_grades)]
names(grade_colors) <- tumor_grades
# 颜色
laterality_types <- unique(data$laterality)
laterality_colors <- brewer.pal(min(length(laterality_types), 9), "Pastel1")[1:length(laterality_types)]
names(laterality_colors) <- laterality_types
# 年龄颜色
age_groups <- unique(data$age)
age_colors <- brewer.pal(min(length(age_groups), 9), "Pastel2")[1:length(age_groups)]
names(age_colors) <- age_groups
# 将所有颜色组合成一个列表
annotation_colors <- list(
Risk_Group = risk_colors,
FIGO_Stage = figo_colors,
Tumor_Grade = grade_colors,
Laterality = laterality_colors,
Age_Group = age_colors
)
# 4. 绘制热图 ----------------------------------------------------------------
# 设置输出文件(如果需要保存到文件)
# pdf("heatmap.pdf", width = 12, height = 8)
# 或者
# png("heatmap.png", width = 1200, height = 800, res = 150)
# 绘制热图
pheatmap(
mat = expression_matrix,
# 注释信息
annotation_col = annotation_col,
annotation_colors = annotation_colors,
# 颜色设置(基因表达量的颜色)
color = colorRampPalette(c("blue", "white", "red"))(100),
# 标准化:对行(基因)进行标准化,使比较更公平
scale = "row",
# 显示设置
show_colnames = TRUE, # 显示列名(样本ID)
show_rownames = TRUE, # 显示行名(基因名)
# 字体大小
fontsize_row = 10,
fontsize_col = 8,
# 图注设置
legend = TRUE,
legend_breaks = c(-2, 0, 2),
legend_labels = c("Low", "Medium", "High"),
# 边界
border_color = NA,
# 聚类设置
cluster_cols = TRUE, # 对列(样本)进行聚类
cluster_rows = TRUE, # 对行(基因)进行聚类
# 树状图设置
treeheight_row = 50,
treeheight_col = 50,
# 单元格大小
cellwidth = ifelse(ncol(expression_matrix) > 30, NA, 15),
cellheight = 15,
# 标题
main = "Gene Expression Heatmap by Risk Group and Clinical Variables",
# 颜色键设置
annotation_legend = TRUE,
annotation_names_col = TRUE,
annotation_names_row = TRUE
)
# 关闭图形设备(如果保存到文件)
# dev.off()
# 5. 可选:简化版热图(只显示风险分组) ----------------------------------------
# 如果所有临床变量太多,可以只展示风险分组
simple_annotation <- data.frame(
Risk_Group = factor(data$risk, levels = c("low risk", "high risk")),
row.names = data$X
)
simple_colors <- list(Risk_Group = risk_colors)
pheatmap(
mat = expression_matrix,
annotation_col = simple_annotation,
annotation_colors = simple_colors,
color = colorRampPalette(c("blue", "white", "red"))(100),
scale = "row",
show_colnames = TRUE,
show_rownames = TRUE,
fontsize_row = 10,
fontsize_col = 8,
main = "Gene Expression Heatmap by Risk Group",
cluster_cols = TRUE,
cluster_rows = TRUE
)
# 6. 可选:按风险组排序的版本 -------------------------------------------------
# 按风险组排序样本
risk_order <- order(data$risk)
expression_matrix_sorted <- expression_matrix[, risk_order]
# 重新创建按排序后的样本顺序的注释
annotation_sorted <- data.frame(
Risk_Group = factor(data$risk[risk_order], levels = c("low risk", "high risk")),
row.names = data$X[risk_order]
)
# 绘制按风险组排序的热图(不聚类)
pheatmap(
mat = expression_matrix_sorted,
annotation_col = annotation_sorted,
annotation_colors = simple_colors,
color = colorRampPalette(c("blue", "white", "red"))(100),
scale = "row",
show_colnames = TRUE,
show_rownames = TRUE,
fontsize_row = 10,
fontsize_col = 8,
main = "Gene Expression Heatmap (Sorted by Risk Group)",
cluster_cols = FALSE, # 不聚类,保持排序顺序
cluster_rows = TRUE
)
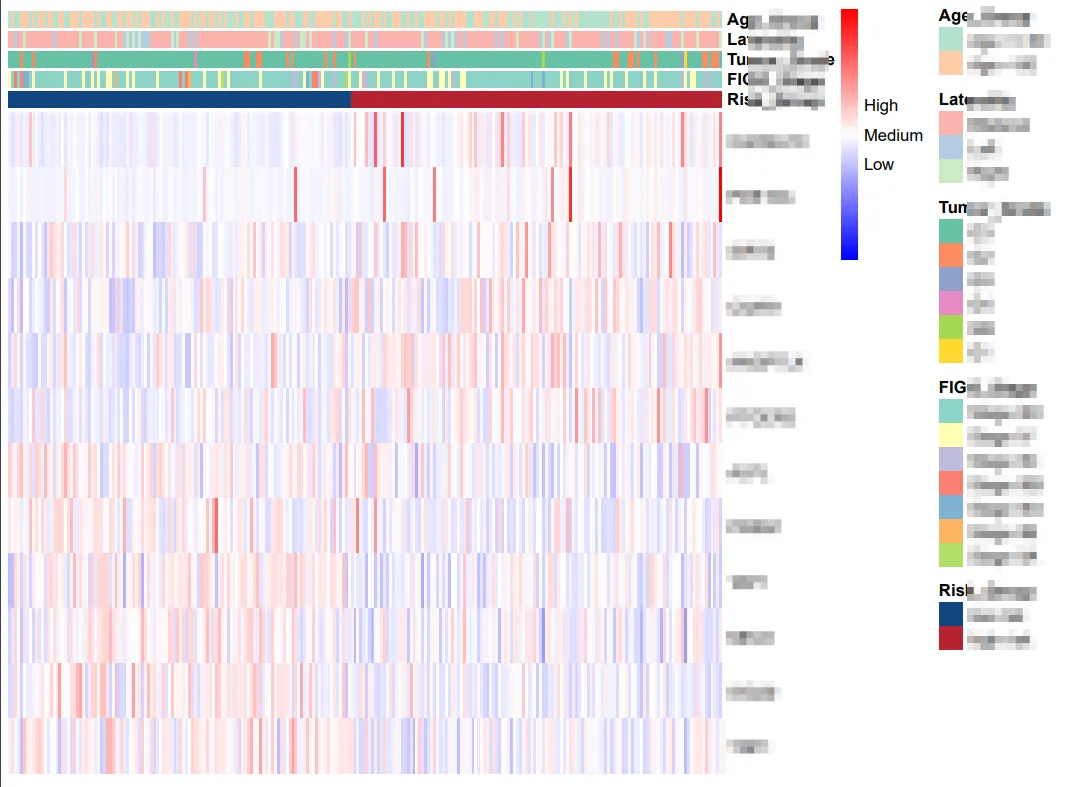
三、AI生成代码的四大亮点
1. 完整的解决方案
AI不仅提供了核心的热图绘制代码,还包括:
数据预处理和提取 多维度临床注释的创建 智能颜色分配系统 多种可视化选项
2. 专业级的细节处理
自动为分类变量分配区分度明显的颜色 对基因表达量进行行标准化(Z-score),确保可比性 提供聚类分析选项,发现潜在模式 完整的图例和标签系统
3. 灵活的定制选项
AI还贴心地提供了三个版本:
完整版:包含所有临床变量的注释 简化版:仅展示风险分组 排序版:按风险组排序,不进行聚类
4. 即用型代码
生成的代码包含详细注释,即便是R语言初学者也能理解每一步的作用。更重要的是——这段代码可以直接运行!
四、从对话到成果:AI理解了什么?
这次交互展示了AI在生物信息学辅助方面的强大能力:
数据结构理解:AI正确识别了数据框结构,提取了正确的基因列和临床信息
可视化需求翻译:将自然语言描述准确转化为技术参数:
“横坐标为不同的分组情况”→ annotation_col参数“颜色按不同分组确定”→ annotation_colors参数“纵坐标为基因”→提取指定基因,转置矩阵 专业知识应用:
使用 pheatmap这一生物信息学常用包默认进行行标准化,这是基因表达热图的常规操作 使用红-蓝渐变色,符合表达量高低的可视化惯例 完整性思维:不仅提供核心功能,还添加了图例、标题、字体调整等细节
五、AI辅助生信绘图的三大优势
🚀 效率革命
从需求描述到获得可运行代码,整个过程仅需几分钟。相比传统的手动编码方式,效率提升超过10倍。
📚 学习加速
对于生信初学者,通过阅读AI生成的结构清晰、注释完整的代码,可以快速学习:
特定分析的标准流程 常用R包的核心函数 可视化最佳实践
🔧 减少错误
AI生成的代码通常遵循良好的编程实践,减少了因语法错误、参数错误导致的调试时间。
六、实践建议:如何更好地与AI协作
提供清晰的数据结构:像示例中那样展示
head(data)的输出,让AI了解你的数据框结构明确具体需求:说明横坐标、纵坐标、颜色映射分别对应什么
指定偏好设置:如果你有特定的颜色偏好、聚类需求等,可以在提示词中说明
迭代优化:如果第一次的结果不完全符合预期,可以继续对话进行微调
七、不止于热图:AI能做的还有更多
这只是AI在生物信息学辅助中的冰山一角。同样的方法可以应用于:
生存分析曲线(Kaplan-Meier plot) 火山图(差异表达分析) 富集分析气泡图 免疫浸润分析图表 基因组浏览器视图
写在最后
AI不是要取代生物信息学家,而是成为我们强大的协作者。它将我们从重复性、机械性的编码工作中解放出来,让我们能够更专注于科学问题的本质——假设提出、结果解读和生物学意义的挖掘。
下一次当你面对数据可视化需求时,不妨尝试与AI对话。你可能会惊喜地发现,那些曾经需要数小时完成的工作,现在只需要一次清晰的对话。
让AI处理代码,让你专注于科学。
以上就是本期全部内容,祝大家学习愉快,收获满满,请给稳妥少年点一个赞、并转发给新的小伙伴吧!!!
稳妥少年建立了第一个讨论群(稳妥少年_共勉学习交流群),特此欢迎新老朋友进群交流学习,让学习变简单!
交流已超200+,无法直接扫码加入,大家如果需要的可以添加微信,拉您进群的。添加微信时,一定要注明来意哈。
稳妥少年——单细胞分析实战
本课程系统介绍了单细胞RNA测序数据分析的全流程方法学体系。从原始数据读取开始,详细讲解了三种常见格式(标准10X输出、HDF5文件和稀疏矩阵)的处理策略,并统一转换为Seurat对象进行分析。质控阶段通过检测基因数、UMI总数和线粒体基因百分比等指标过滤低质量细胞,确保数据可靠性。标准化环节采用对数归一化和高变基因识别方法,通过主成分分析降维,使用JackStraw检验和肘部图确定最佳维度。
细胞聚类分析采用共享最近邻算法和Louvain聚类,在UMAP降维空间可视化细胞亚群。基于经典标记基因表达模式,对细胞类型进行系统注释。差异表达分析采用多层次策略,包括整体水平比较、细胞类型特异性分析和功能富集评估。细胞比例分析通过统计检验评估实验组间细胞组成的差异。
高级分析模块涵盖细胞间通讯网络构建(CellChat)、发育轨迹推断(Monocle)、代谢通路活性评估(scMetabolism)以及基因集富集分析(GSEA)。特别整合了GSVA通路活性评分、AddModuleScore模块化评分等创新方法,全面解析细胞功能状态。可视化方面采用多类型图表(火山图、热图、轨迹图、网络图等)呈现分析结果,所有方法均经过标准化流程验证,确保分析结果的可靠性和可重复性,为疾病机制研究和生物标志物发现提供系统解决方案。
【淘宝】限时满500减200 https://e.tb.cn/h.73SuB1NblkWjvxx?tk=QJ1vfvkGE3A HU293 「稳妥少年,单细胞分析实战课」
点击链接直接打开 或者 淘宝搜索直接打开
咸鱼
【闲鱼】https://m.tb.cn/h.73SGjJy?tk=tq8efvkucB3 HU071 「我在闲鱼发布了【【稳妥少年-单细胞分析实战全流程视频课】】」
点击链接直接打开
稳妥少年——生信技能课
本课程系统涵盖了生物信息学在基因组数据挖掘与分析中的核心流程与方法。课程从GEO、TCGA等公共数据库的数据检索与下载入门,进而学习差异表达分析(使用limma、DESeq2等工具)及结果可视化。在此基础上,深入进行功能富集分析(GO、KEGG)和蛋白质互作网络构建,并利用Cytoscape筛选关键基因模块。课程还整合了多种机器学习算法进行生物标志物筛选,并教授诊断列线图、神经网络模型等临床预测模型的构建与评估。此外,课程内容扩展至基因集富集分析(GSEA/GSVA)、免疫细胞浸润分析、基因的亚细胞与染色体定位、表型关联分析、表达相关性分析、单基因预后分析以及肿瘤免疫检查点分析等一系列高级专题,旨在提供从基础到前沿的完整生物信息学分析能力。
淘宝
【淘宝】限时满500减200 https://e.tb.cn/h.7X8RTTPSng5IcEN?tk=iJN5fGQL1mn MF278 「稳妥少年一生信技能课」
点击链接直接打开 或者 淘宝搜索直接打开
咸鱼
【闲鱼】https://m.tb.cn/h.72Tg8o6?tk=F34xfGQNQAt HU293 「我在闲鱼发布了【本课程《生物信息学全流程实战技能课》系统讲授疾病研究,尤其是】」
点击链接直接打开
TCGA肿瘤预后+单细胞+虚拟基因敲除
本研究系统整合了多组学数据与生物信息学方法,旨在探究多发性骨髓瘤的分子特征、预后标志及免疫微环境。首先,从TCGA与GEO数据库获取并标准化了批量转录组数据,通过差异表达分析鉴定了疾病相关的关键基因,并利用GO/KEGG富集分析阐释其功能。基于此,采用Cox回归与LASSO算法构建并验证了一个多基因预后风险模型,该模型在独立队列中展现出稳健的预测效能。进一步分析揭示了风险评分与临床病理特征的独立性,并通过列线图实现了个体化生存预测。
为深入解析肿瘤微环境,本研究应用ssGSEA评估了免疫细胞浸润水平,并分析了其与风险模型及免疫检查点表达的相关性。同时,利用单细胞RNA测序技术,在细胞分辨率下刻画了骨髓瘤微环境的细胞组成图谱,识别了主要免疫细胞亚群,并量化了其在不同临床状态下的比例变化。通过CellChat工具,系统推断了疾病组与对照组间细胞通讯网络的差异。此外,采用scTenifoldKnk算法对髓系细胞亚群进行了虚拟基因敲除分析,从网络层面探索了关键基因的调控功能。
综上,本研究通过从宏观群体水平到微观单细胞层面的多层次分析,构建了多发性骨髓瘤的预后预测体系,并深入揭示了其肿瘤免疫微环境的特征与潜在调控机制,为理解疾病生物学及开发治疗策略提供了新的见解和数据支持。

淘宝
【淘宝】https://e.tb.cn/h.74PYbf9cYTjXRw9?tk=o9KTfxHXS3R CZ005 「稳妥少年【肿瘤预后+单细胞+基因敲除】全流程视频课」
点击链接直接打开 或者 淘宝搜索直接打开
咸鱼
【闲鱼】https://m.tb.cn/h.7fqO1d0?tk=KEbifxHcGH5 CA381 「我在闲鱼发布了【肿瘤预后+单细胞+基因敲除全流程视频课】」
点击链接直接打开
稳妥少年【NHANES数据库使用教程】
这门课是面向临床研究的“流水线”实战培训,核心是教你把NHANES数据库的原始文件变成一张张能用于论文的图表和表格。它会手把手带你走通全流程:从用R代码自动下载、合并不同年份的数据模块,到严格清洗变量、筛选研究对象;从正确运用复杂的抽样权重进行基线统计与加权逻辑回归,到使用限制性立方样条(RCS)探索变量间的非线性关系;最后,指导你完成单因素/多因素分析,并绘制出可直接用于发表的森林图。整个过程紧扣实际研究需求,重点解决如何正确处理权重、合并数据、校正混杂因素等具体技术难题,目标是让你能独立完成一项从数据到结果的完整分析。
淘宝
【淘宝】https://e.tb.cn/h.7WtLo3unmwB0dUI?tk=7XlqfEbwdDS MF937 「稳妥少年—NHANES数据库使用 全流程视频课」
点击链接直接打开 或者 淘宝搜索直接打开
咸鱼
【闲鱼】https://m.tb.cn/h.73y4RJk?tk=yhJffEbvOgU MF287 「我在闲鱼发布了【稳妥少年—NHANES数据库使用】」
点击链接直接打开
稳妥少年网络毒理学课程
网络毒理学分析流程详细内容
(1)疾病转录组数据准备(2)毒物毒理学评估(3)毒物靶点基因的获取(4)疾病靶点基因的获取(5)差异表达基因的获取(6)候选基因的获取(7)GO与KEGG富集分析(8)毒物-基因-通路-疾病网络构建(9)蛋白质相互作用网络构建和核心基因筛选(10)表达水平验证筛选关键基因(11)相关性和功能相似性分析(12)转录因子-基因-miRNA调控网络构建(13)关键基因富集分析(14)GeneMANIA分析、亚细胞定位和染色体定位分析(15)分子对接(16)分子动力学模拟

课程购买链接
淘宝
【淘宝】https://e.tb.cn/h.SyH6B9i86ptdeNZ?tk=MiTYfLWdlC2 CZ005 「稳妥少年【网络毒理学全流程视频课】」
点击链接直接打开 或者 淘宝搜索直接打开
咸鱼
【闲鱼】https://m.tb.cn/h.SyhI8J3?tk=1BqmfLWUyIH CZ028 「我在闲鱼发布了【稳妥少年【网络毒理学全流程视频课】】」
点击链接直接打开
分子对接、动力学模拟课程推荐
课程一:《分子对接零基础教学课程上线 | ADT/Vina/Maestro全流程详解》
介绍:
本课程系统讲解从原理到实操的全流程操作,配套三大常用工具教学,助你快速掌握药物设计核心技能。
课程主要内容包括:分子对接原理、全流程梳理、软件下载、蛋白质小分子数据库介绍、ADT受配体结果预处理、对接口袋预测及设置、vina\qvina分子对接计算、结果可视化相互作用力分析、Maestro软件安装介绍、Maestro分子对接计算、高通量分子对接、Maestro蛋白质-蛋白质对接计算
咸鱼平台购买链接
【闲鱼】https://m.tb.cn/h.6lrKXb4?tk=EaHSVelRlCx HU293 「我在闲鱼发布了【【分子对接零基础教学课程上线 | ADT/Vina/Maes】」
点击链接直接打开
淘宝购买链接:
【淘宝】https://e.tb.cn/h.6lrv1KbfzG5szI9?tk=KQ3OVelmPFZ CZ193 「【分子对接零基础教学课程上线 | ADT/Vina/Maestro全流程详解】」
点击链接直接打开 或者 淘宝搜索直接打开
课程二:《分子动力学全流程视频课程》
介绍:
本课程系统讲解从原理到实操的全流程操作,配套三大常用工具教学,助你快速掌握药物设计核心技能。
课程总时长4小时左右,从动力学模拟原理解读、GROMACS软件安装、受配体拆分预处理、拓扑文件构建、模拟体系搭建、动力学模拟以及下游分析、结果图的绘制和解读、文章发表的撰写方法;可以说是全流程一课通!!!
咸鱼平台购买链接
【闲鱼】https://m.tb.cn/h.69TGRAA?tk=bzDYVde3bcC HU293 「我在闲鱼发布了【稳妥少年,从2022年开始主要涉及分子对接和分子动力学模拟领】」
点击链接直接打开
淘宝购买链接:
【淘宝】https://e.tb.cn/h.69hWyklEydyhI5G?tk=hvzpVdVOI6F HU293 「稳妥少年《分子动力学全流程视频课程》时长4小时左右,一课通!」
点击链接直接打开 或者 淘宝搜索直接打开
优惠活动:现在同时购买两套课程,赠送稳妥少年自研的《高通量分子对接工具HTMDv3》,赠送9113个小分子库,以及2101个重要活性分子库,助力大家进行药研相关工作!!!
工具介绍:高通量分子对接软件(HTMDv3)介绍及教学——稳妥少年

祝大家学习愉快!也感谢大家对公众号的支持,继续努力!!!今天分享的内容可能存在不足,希望感兴趣的小伙伴进行批评指正。同时也希望大家可以一键三连(点赞、关注和转发“稳妥少年”)