在数据科学和化学信息学(Cheminformatics)领域,机器学习正在发挥着越来越重要的作用。今天,我们将手把手带你走过一个完整的机器学习实战项目!本文将从最基础的 Scikit-learn (Sklearn) 用法讲起,一步步深入到真实的药物分子数据清洗、特征提取(RDKit)、模型构建、超参数优化以及模型保存。所有代码已整理完毕,建议打开 Jupyter Notebook 跟着敲一遍!
零、 准备工作:环境搭建
在开始之前,请确保你已经安装了以下关键的 Python 库:
Bash
pip install --upgrade scikit-learn pandas scipy joblib# 建议通过 conda 安装 RDKitconda install -c conda-forge rdkitpip install standardiser
一、 Sklearn 基础热身:模型训练与属性探索
在处理复杂的化学数据之前,我们先用经典的开源数据集熟悉一下 Sklearn 的核心 API。Sklearn 的设计非常优雅,几乎所有模型都遵循 fit() (训练) 和 predict() (预测) 的模式。
1. K近邻分类器 (KNN) 与鸢尾花数据集
通过计算样本之间的距离来进行分类,是非常直观的基础算法。
Python
from sklearn import datasetsfrom sklearn.model_selection import train_test_splitfrom sklearn.neighbors import KNeighborsClassifier# 加载鸢尾花数据集iris = datasets.load_iris()iris_X = iris.datairis_y = iris.target# 拆分数据集:70%用于训练,30%用于测试X_train, X_test, y_train, y_test = train_test_split(iris_X, iris_y, test_size=0.3)# 构建KNN模型并训练knn = KNeighborsClassifier()knn.fit(X_train, y_train)# 进行预测print("预测结果:", knn.predict(X_test))print("真实结果:", y_test)
2. 线性回归与模型属性探索
模型训练好之后,内部会保存很多“学习到”的参数。我们以波士顿房价数据集(或类似回归数据)为例,看看如何提取这些属性:
Python
from sklearn import datasetsfrom sklearn.linear_model import LinearRegression# 引入数据load_data = datasets.load_boston()data_X = load_data.datadata_y = load_data.targetprint("数据维度:", data_X.shape)# 构建并训练线性回归模型model = LinearRegression()model.fit(data_X, data_y)# 预测前4条数据print("预测前4条数据:", model.predict(data_X[:4, :]))# 探索 Sklearn 模型的内置属性和功能print("模型系数 (权重):", model.coef_)print("模型截距:", model.intercept_)print("模型参数:", model.get_params())print("模型R^2评分:", model.score(data_X, data_y))
3. 数据预处理:标准化 (Standardization)
真实世界的数据尺度往往差异巨大。为了防止大数值特征主导模型,我们需要对数据进行缩放处理,使其均值为 0,方差为 1。
Python
from sklearn import preprocessingimport numpy as npa = np.array([[10, 2.7, 3.6], [-100, 5, -2], [120, 20, 40]], dtype=np.float64)print("原始数据:\n", a)# 数据标准化print("标准化后的数据:\n", preprocessing.scale(a))
二、 进阶实战:化学分子数据的预处理
1.下载数据集
打开https://pubchem.ncbi.nlm.nih.gov/,搜索框搜索1851,拉到BIOASSAY,找到AID为1851的,点击打开接下来,我们进入真实的业务场景:预测小分子化合物的性质(例如 CYP1A2 酶抑制活性)。我们将使用 pandas 处理表格,使用 RDKit 和 standardiser 处理化学结构。
2. 读取数据与分子结构标准化
我们从 cyp1a2.csv 中读取包含 SMILES 序列的数据,对其进行脱盐、标准化等操作,以确保输入给模型的分子结构是统一且正确的。
Python
import pandas as pdimport numpy as npimport loggingfrom rdkit import Chem from rdkit.Chem import Descriptorsfrom standardiser import standardise# 读取并清理空值df = pd.read_csv('cyp1a2.csv')df = df.dropna()# 遍历数据,进行 SMILES 标准化for i in df.index:try: smi = df.loc[i, 'SMILES'] mol = Chem.MolFromSmiles(smi) mol = Chem.AddHs(mol) # 添加氢原子 parent = standardise.run(mol) # 结构标准化 mol_ok_smi = Chem.MolToSmiles(parent) df.loc[i, 'SMILES'] = mol_ok_smiexcept standardise.StandardiseException as e: logging.warning(e.message)# 去除重复项df.drop_duplicates(keep='first', inplace=True)
2. 计算分子描述符与数据过滤
我们利用 RDKit 计算分子的物理化学性质(如分子量 MolWt 和脂水分配系数 LogP),并过滤掉异常庞大的分子。
Python
molweight = []logP = []for smi inlist(df['SMILES']): mol = Chem.MolFromSmiles(smi) molweight.append(Descriptors.MolWt(mol)) logP.append(Descriptors.MolLogP(mol))# 将计算结果存入 DataFramedf['molecular_weight'] = molweightdf['logP'] = logP# 过滤掉分子量大于1000的宏大分子df = df[df['molecular_weight'] <= 1000]# 保存清洗后的高质量数据df.to_csv('data.csv', index=None)print(df.head())
三、 特征工程与建模评估
机器学习模型看不懂化学结构图,我们需要将 SMILES 转化为模型能理解的“分子指纹”(如 Morgan Fingerprint 和 MACCS keys)。
1. 核心库导入解析
在此阶段,我们会用到大量 Sklearn 工具。先来理解它们的作用:
KNeighborsClassifier / SVC / RandomForestClassifier: 各种分类算法(KNN、支持向量机、随机森林)。
train_test_split: 划分训练集和测试集。
GridSearchCV: 超参数优化神器,通过交叉验证自动寻找最佳参数组合。
roc_auc_score / confusion_matrix: 模型性能评估工具。
2. 方案 A:Morgan 指纹 + KNN 模型优化
我们将生成 Morgan 指纹(长度为 1024 位的二进制向量),并结合 KNN 算法与网格搜索(GridSearchCV)寻找最佳的 n_neighbors。
Python
from rdkit.Chem import AllChemfrom sklearn.neighbors import KNeighborsClassifierfrom sklearn.model_selection import train_test_split, GridSearchCVfrom sklearn.metrics import roc_auc_score, confusion_matriximport mathdf = pd.read_csv('data.csv')# 1. 生成 Morgan 分子指纹作为特征 XX = np.array([AllChem.GetMorganFingerprintAsBitVect(Chem.MolFromSmiles(smi), 2, nBits=1024) for smi inlist(df['SMILES'])])y = df['Label'].values# 2. 拆分数据集X_train, X_test, y_train, y_test = train_test_split(X, y, random_state=0)# 3. 构建KNN模型并使用交叉验证优化超参数knn = KNeighborsClassifier()param = {"n_neighbors": [3, 5, 10]}gc_knn = GridSearchCV(knn, param_grid=param, cv=5, scoring='roc_auc')gc_knn.fit(X_train, y_train)print("KNN 最佳参数组合:", gc_knn.best_params_)# 4. 预测与评估y_pred = gc_knn.predict_proba(X_test)[:, 1] # 获取正类的预测概率auc_score = roc_auc_score(y_test, y_pred)print("ROC-AUC 评分:", auc_score)
3. 方案 B:MACCS 密钥 + SVM 模型优化
我们再尝试另一种指纹 MACCS,并使用支持向量机(SVC)。
Python
from rdkit.Chem import MACCSkeysfrom sklearn.svm import SVC# 1. 生成 MACCS keys 作为特征 XX = np.array([MACCSkeys.GenMACCSKeys(Chem.MolFromSmiles(smi)) for smi inlist(df['SMILES'])])y = df['Label'].valuesX_train, X_test, y_train, y_test = train_test_split(X, y, random_state=0)# 2. 构建模型并定义超参数优化空间svc = SVC(probability=True)param = {"gamma": ['scale', 'auto', 0.1], 'C': [1, 10, 100]}gc_svc = GridSearchCV(svc, param_grid=param, cv=5, scoring='roc_auc')gc_svc.fit(X_train, y_train) # 注意:如果是大数据集,建议使用 X_train 进行拟合print("SVC 最佳参数组合:", gc_svc.best_params_)
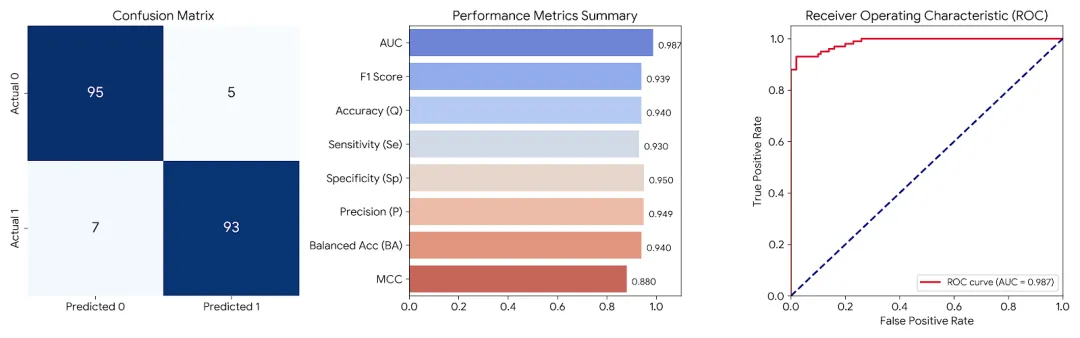
四、 模型评估:硬核指标全解析
有了预测结果 y_pred 和真实标签 y_test,我们如何判断模型到底行不行?以下是必备的评估指标计算全过程:
Python
# 假设我们使用上面跑完的 gc_svc 模型结果y_pred = gc_svc.predict_proba(X_test)[:, 1]auc_roc_score = roc_auc_score(y_test, y_pred)# 将概率转为 0 或 1 的硬标签用于计算混淆矩阵y_pred_print = [round(y, 0) for y in y_pred]tn, fp, fn, tp = confusion_matrix(y_test, y_pred_print).ravel()# 计算各项指标se = tp / (tp + fn)sp = tn / (tn + fp)q = (tp + tn) / (tp + fn + tn + fp)mcc = (tp * tn - fn * fp) / math.sqrt((tp + fn) * (tp + fp) * (tn + fn) * (tn + fp))P = tp / (tp + fp)F1 = (P * se * 2) / (P + se)BA = (se + sp) / 2print("TP:", tp, "TN:", tn, "FP:", fp, "FN:", fn)print(f"灵敏度(Se): {se:.3f}, 特异性(Sp): {sp:.3f}, 准确率(Q): {q:.3f}")print(f"MCC: {mcc:.3f}, AUC: {auc_roc_score:.3f}, F1: {F1:.3f}, BA: {BA:.3f}"
📊 核心指标原理解析:
TP / TN / FP / FN:真正例、真负例、假正例、假负例。这是所有指标的基础构件。
Sensitivity (Se,灵敏度/召回率):$Se = \frac{TP}{TP + FN}$。模型正确识别正类(如:有活性的分子)的比例。
Specificity (Sp,特异性):$Sp = \frac{TN}{TN + FP}$。模型正确识别负类的比例。
Accuracy (Q,准确率):$Q = \frac{TP + TN}{TP + FN + TN + FP}$。模型整体猜对的比例。
MCC (马修斯相关系数):$MCC = \frac{TP \cdot TN - FP \cdot FN}{\sqrt{(TP + FN)(TP + FP)(TN + FN)(TN + FP)}}$。衡量分类的相关性,范围 [-1, 1]。1 为完美分类,0 为随机,面对样本不平衡时非常有效。
Precision (P,精确率):$P = \frac{TP}{TP + FP}$。预测出来的正类中,有多少是真的正类。
F1 Score:$F1 = \frac{2 \cdot P \cdot Se}{P + Se}$。精确率和召回率的调和平均数。
AUC-ROC Score:最权威的区分能力指标,范围 [0.5, 1]。越接近 1 模型性能越优异。
(注:如果你的模型跑出高灵敏度、高特异性,且 AUC 达到 0.90 以上,说明你的模型具有非常优秀的分子活性分类能力!)
五、 模型持久化:保存与加载
训练一个优质模型耗时耗力,测试完成后,我们必须要将它保存下来,方便日后直接调用或部署。Python 提供了多种保存方法:
1. 使用内置的 Pickle 库
Python
import pickleimport osfrom sklearn import svm, datasetsos.makedirs('save', exist_ok=True) # 创建目录# 训练一个简单的模型作为示例df_model = svm.SVC()iris = datasets.load_iris()X, y = iris.data, iris.targetdf_model.fit(X, y)# 保存模型 (写入二进制序列化文件)withopen('save/clf.pickle', 'wb') as f: pickle.dump(df_model, f)# 加载模型并进行预测withopen('save/clf.pickle', 'rb') as f: clf_loaded = pickle.load(f) print("Pickle 加载预测结果:", clf_loaded.predict(X[0:1]))
2. 使用 Joblib (推荐)
对于 Sklearn 中包含大量 Numpy 数组的模型,joblib 库的效率远高于 pickle。
Python
import joblib# 保存模型joblib.dump(df_model, 'save/clf.pkl')# 加载模型并预测model_joblib = joblib.load('save/clf.pkl')print("Joblib 加载预测结果:", model_joblib.predict(X[0:1]))
🎉 总结
恭喜你!到这里,我们已经走过了一个基于 Scikit-learn 和 RDKit 的机器学习完整生命周期。从数据标准化的细致操作,到超参数优化的精益求精,再到最后硬核的模型指标评估,你已经掌握了 AI 药物研发数据科学家的必备技能!
赶紧打开你的代码编辑器,把文章里的代码跑通一遍吧!