编者按
我们推出了一个新的栏目,重构算法的介绍,我们将介绍omicverse开源重构生态最新的成果以及完整的教程,欢迎关注。
本期教程里,我们给大家带来的是单细胞TCR免疫组库分析:这是omicverse中 ov.airr 模块的内容。我们在omicverse中整合了单细胞TCR分析常用的一套流程,包括TCR数据读入、chain QC、clonotype定义、克隆扩增、clonotype network、repertoire diversity、V(D)J gene usage、MAIT/iNKT识别、clonotype imbalance、转录状态一致性、CDR3 logo以及VDJdb抗原注释。
本文使用Wu 2020肿瘤T细胞单细胞TCR-seq + RNA-seq,演示如何用OmicVerse完成一套完整的单细胞免疫组库分析。
本文AI率20%,阅读大约需要16min。
OmicVerse团队
1. 引言
T细胞有两套身份。
第一套身份来自转录组。它告诉我们这个T细胞处在什么状态:naive、effector、exhausted、Treg、Trm,或者其他更细的亚群。
第二套身份来自TCR。TCR在V(D)J重排时形成,后续克隆扩增时会被复制到所有子代细胞中。因此,如果一群T细胞拥有相同TCR,我们通常可以认为它们来自同一个克隆。
这就是单细胞TCR分析最有价值的地方。
如果只做scRNA-seq,我们能看到T细胞状态,但不知道哪些细胞来自同一个克隆扩增。
如果只做bulk TCR,我们能看到克隆扩增,但不知道这些克隆到底是Teff、Trm、Treg还是naive-like。
单细胞TCR + 转录组联合分析,可以直接回答一个更有意思的问题:
哪些转录状态里的T细胞正在发生克隆扩增?
这在肿瘤免疫里很有用。因为真正可能识别肿瘤抗原的T细胞,往往不会随机分布在所有T细胞状态中,它们更容易集中在某些CD8 effector、Trm或exhausted-like状态里。
本期教程使用Wu 2020的单细胞TCR数据,跑一遍从TCR QC到VDJdb抗原注释的完整流程。
2. 数据读入
首先载入OmicVerse并设置绘图风格:
import omicverse as ov
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
ov.plot_set()
读取示例数据:
adata = ov.datasets.airr_singlecell()
print(f"matrix: {adata.n_obs} cells x {adata.n_vars} genes")
print(f"raw counts in .X — max value {adata.X.max():.0f}")
print(f"obs columns : {list(adata.obs.columns)}")
print(f"obsm keys : {list(adata.obsm.keys())}")
matrix: 5001 cells x 13968 genes
raw counts in .X — max value 9493
obs columns : ['patient', 'sample', 'source', 'cluster_orig', 'cell_type',
'clonotype_orig', 'high_confidence', 'is_cell']
obsm keys : ['X_umap_orig', 'airr']
这个数据包含5001个T细胞和13968个基因。
adata.X是基因表达矩阵,adata.obsm['airr']里存的是每个细胞对应的TCR contigs。也就是说,这个AnnData对象同时带着转录组和TCR信息。
看一下样本来源:
for c in ["source", "patient"]:
print(f"--- {c} ---")
print(adata.obs[c].value_counts())
--- source ---
source
Tumor 2594
NAT 2065
Blood 342
--- patient ---
patient
Lung2 707
Lung3 624
Lung6 619
Lung1 541
Endo1 520
Lung4 455
Renal2 357
Lung5 251
Endo2 203
Endo3 198
Renal1 196
Colon1 154
Renal3 151
Colon2 25
这里有三个组织来源:肿瘤、癌旁组织和外周血。
先看作者提供的转录组UMAP:
ov.pl.embedding(
adata,
basis="X_umap_orig",
color="cell_type",
frameon="small",
title="Transcriptome states (Wu 2020)",
show=False,
)
plt.show()
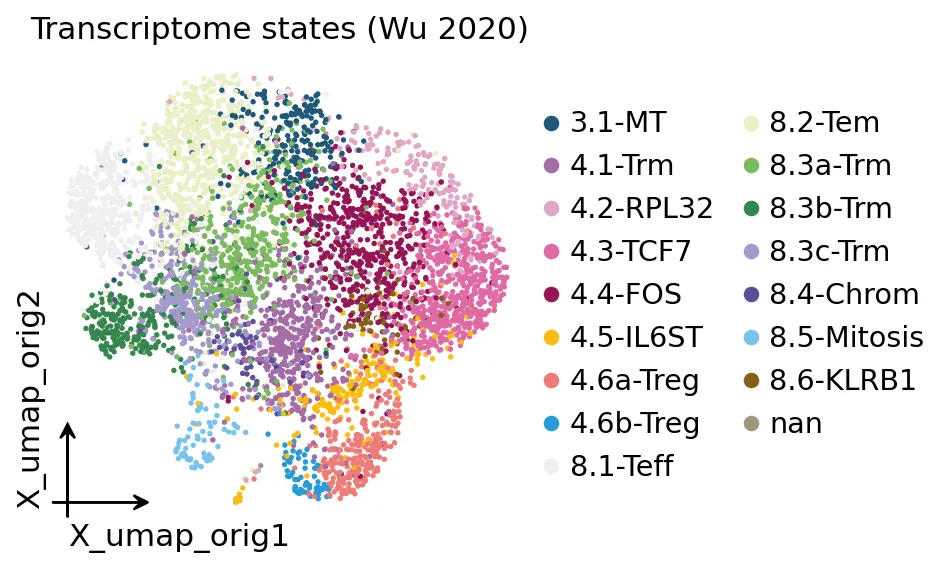 Wu 2020 T细胞转录组UMAP
Wu 2020 T细胞转录组UMAP这张图里,每个点是一个T细胞,颜色是作者给出的细胞状态注释。
后面我们要做的事情,就是把TCR克隆信息放回这张转录组的umap图里,看哪些转录状态发生了克隆扩增。
3. 把TCR数据接入ov.airr格式
原始TCR contigs存在 adata.obsm['airr'] 中,它是一个variable-length的结构。每个细胞可能有一条链,也可能有多条链。
为了方便后续分析,我们用 ov.airr.from_airr_array 把这些contigs整理到 adata.obs 中:
adata = ov.airr.from_airr_array(adata)
new_cols = [c for c in adata.obs.columns if c.startswith(("VJ_", "VDJ_"))]
print(f"per-cell AIRR columns added : {len(new_cols)}")
print("receptor type per cell:")
print(adata.obs["receptor_type"].value_counts())
per-cell AIRR columns added : 36
receptor type per cell:
receptor_type
TCR 4691
ambiguous 279
no IR 27
BCR 4
大部分细胞是TCR,少数是ambiguous或者no IR。这里先不急着过滤,我们先做chain QC。
4. Chain QC
真实T细胞一般应该有一条alpha链和一条beta链。
但单细胞数据里会遇到两类问题:
- 有些droplet可能是doublet,出现多条alpha或多条beta链。
因此我们需要先判断每个细胞的TCR链是否可用。
ov.airr.chain_qc(adata)
print(adata.obs["chain_pairing"].value_counts())
print()
print("receptor subtype:")
print(adata.obs["receptor_subtype"].value_counts().head(3))
chain_pairing
single pair 2593
orphan VDJ 1330
multichain 483
no IR 330
orphan VJ 265
receptor subtype:
receptor_subtype
TRA+TRB 4693
ambiguous 277
no IR 27
这里我们保留 single pair、orphan VJ 和 orphan VDJ,去掉 multichain 和 no IR。
usable = ["single pair", "orphan VJ", "orphan VDJ"]
tcr = adata[adata.obs["chain_pairing"].isin(usable)].copy()
n_drop = adata.n_obs - tcr.n_obs
print(f"cells with a usable TCR : {tcr.n_obs} / {adata.n_obs}")
print(f"dropped (multichain / no IR): {n_drop}")
cells with a usable TCR : 4188 / 5001
dropped (multichain / no IR): 813
也就是说,后续分析基于4188个带有可用TCR的细胞。
这一步的意义在于,doublet会制造假的TCR组合,后面定义clonotype时会直接影响结果。
5. 定义clonotype
TCR clonotype一般可以有两种定义。
第一种是严格定义:alpha链和beta链CDR3完全一致的细胞,被定义为同一个clonotype。
第二种是距离定义:CDR3相似到一定程度的细胞,可以合并成clonotype cluster。这个适合找可能识别同一抗原的近似TCR,但对普通克隆扩增分析来说会更宽松。
这里我们先用严格定义:
ov.airr.define_clonotypes(tcr)
n_clono = tcr.uns["clonotype"]["n_clonotypes"]
print(f"exact clonotypes : {n_clono}")
print(f"largest clone : {int(tcr.obs['clone_id_size'].max())} cells")
print(f"cells in a clone of size >= 2: {(tcr.obs['clone_id_size'] >= 2).sum()}")
exact clonotypes : 3348
largest clone : 27 cells
cells in a clone of size >= 2: 1102
再看距离定义:
ov.airr.define_clonotype_clusters(tcr, metric="hamming", cutoff=1)
n_clust = tcr.uns["clonotype_clusters"]["n_clusters"]
print(f"exact clonotypes : {n_clono}")
print(f"distance-based clusters : {n_clust}")
exact clonotypes : 3348
distance-based clusters : 3331
可以看到,允许一个Hamming mismatch以后,clonotype数量只从3348降到3331,差别很小。
这符合TCR的特点。TCR不像BCR那样会发生体细胞高频突变,所以后面我们可以继续使用严格定义的 clone_id。
6. 克隆扩增
定义完clonotype以后,就可以统计每个细胞所属克隆的大小。
ov.airr.clonal_expansion(tcr)
exp = tcr.obs["clonal_expansion"].value_counts()
frac_exp = 100.0 * (tcr.obs["clonal_expansion"] != "1 (single)").mean()
print(exp)
print(f"\ncells in an expanded (>=2) clone: {frac_exp:.1f}%")
clonal_expansion
1 (single) 3010
>= 4 531
2 424
3 147
cells in an expanded (>=2) clone: 28.1%
也就是说,在这4188个可用TCR细胞里,大约28.1%的细胞属于扩增克隆。
按组织来源拆开看:
ov.airr.clonal_expansion_plot(tcr, groupby="source")
plt.show()
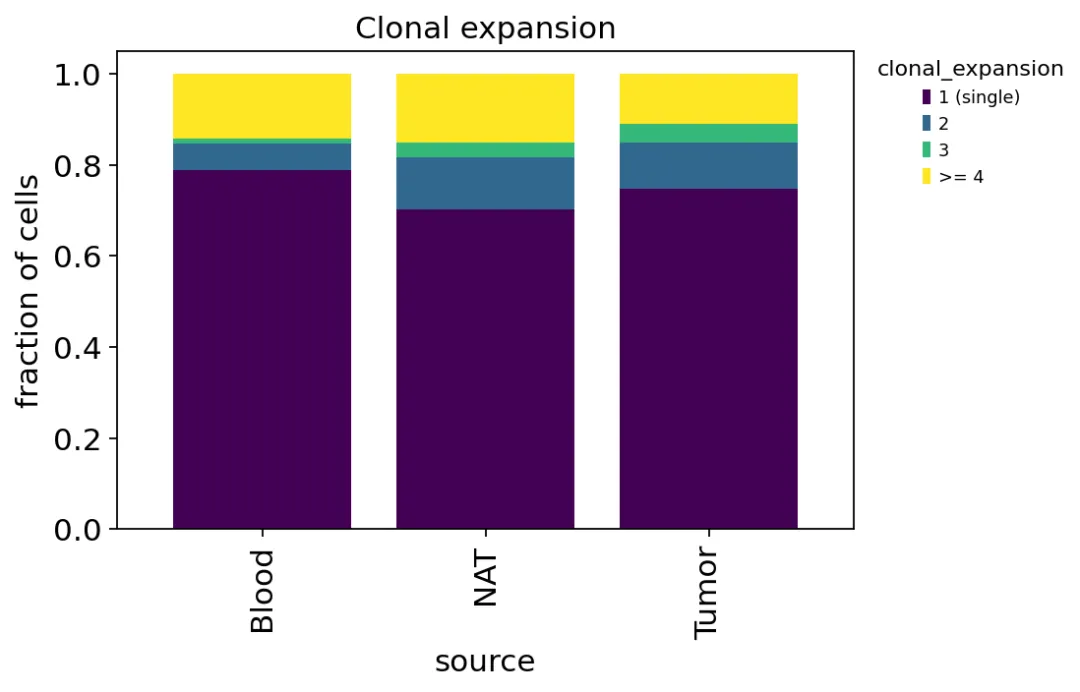 不同组织来源的克隆扩增
不同组织来源的克隆扩增肿瘤里的扩增比例最高,血液里最低。
这其实已经很接近肿瘤TCR分析最想问的问题了:如果某些T细胞在肿瘤里被抗原刺激并增殖,那么它们应该在肿瘤组织里形成更明显的克隆扩增。
7. Clonotype network
Clonotype network可以把扩增克隆画成网络。
每个点是一个细胞,属于同一个clonotype的细胞会连在一起。我们这里只画size >= 2的扩增克隆,否则几千个singleton会把图淹没。
ov.airr.clonotype_network(tcr, min_cells=2)
n_comp = tcr.uns["clonotype_network"]["n_components"]
print(f"expanded clonotypes drawn (size >= 2): {n_comp}")
expanded clonotypes drawn (size >= 2): 338
先按克隆扩增程度上色:
ov.airr.clonotype_network_plot(
tcr,
color="clonal_expansion",
title="Clonotype network (expanded clones)",
)
plt.show()
 按克隆扩增程度上色的clonotype network
按克隆扩增程度上色的clonotype network每一个环就是一个扩增克隆。越大的环,说明这个clone里的细胞越多。
再按组织来源上色:
ov.airr.clonotype_network_plot(
tcr,
color="source",
title="Clonotype network by tissue",
)
plt.show()
 按组织来源上色的clonotype network
按组织来源上色的clonotype network这张图很有意思。很多扩增克隆里同时包含Tumor和NAT细胞,少数还会包含Blood细胞。这说明同一个T细胞克隆可以出现在不同组织来源中,提示T细胞克隆在肿瘤、癌旁组织和外周血之间有共享关系。
8. Repertoire diversity和overlap
我们先看不同组织来源的alpha diversity。
div = ov.airr.alpha_diversity(
tcr,
groupby="source",
metric="normalized_shannon",
)
print(div)
n_cells n_clonotypes normalized_shannon
group
Blood 307 272 0.975210
NAT 1678 1378 0.980015
Tumor 2127 1818 0.984513
再同时计算几个指标:
summary = ov.airr.alpha_diversity(
tcr,
groupby="source",
metric=["normalized_shannon", "gini_simpson", "d50"],
)
summary.round(3)
n_cells n_clonotypes normalized_shannon gini_simpson d50
group
Blood 307 272 0.975 0.993 119
NAT 1678 1378 0.980 0.999 539
Tumor 2127 1818 0.985 0.999 755
这里要稍微注意一下解释。
这几个值都很高,说明三个组织来源里都还有大量singleton。Tumor的细胞数和clonotype数也更多,所以单独看normalized Shannon并不一定能得出“肿瘤多样性下降”的简单结论。
在实际项目中,我更建议把diversity和clonal expansion一起看。肿瘤里确实有更多扩增克隆,但它同时也有大量不同的TCR进入组织,所以整体多样性指标未必会下降。
再看患者之间的repertoire overlap:
ov.airr.repertoire_overlap_plot(tcr, groupby="patient", metric="jaccard")
plt.show()
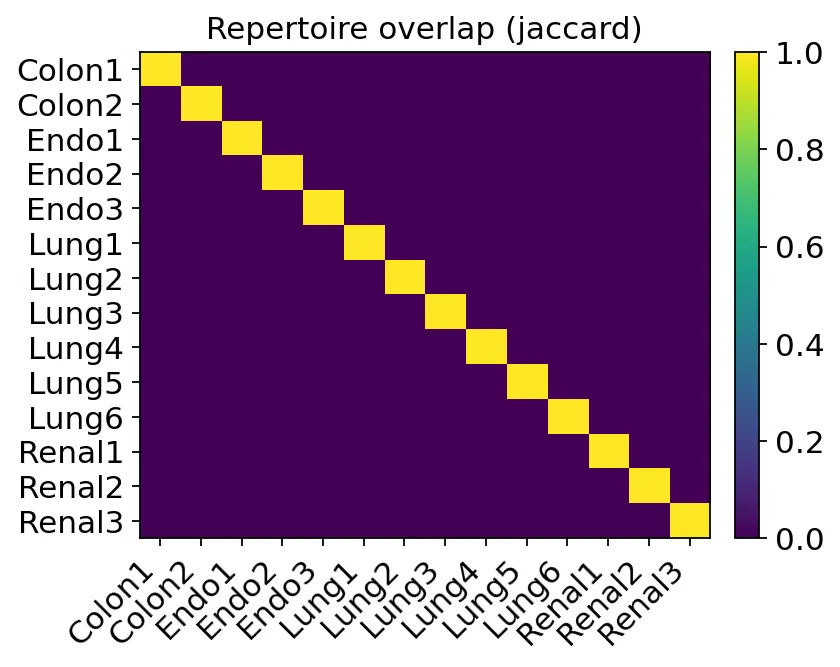 患者间TCR overlap
患者间TCR overlap患者之间的Jaccard overlap几乎接近0。
这是合理的。严格CDR3定义下,大多数TCR clonotype都是patient-private。如果不同患者之间出现大量完全相同的clonotype,那反而要怀疑样本污染、barcode混淆或者定义过宽。
9. V(D)J gene usage
接下来查看TRBV基因使用情况。
ov.airr.vdj_usage_plot(
tcr,
gene="v",
chain="VDJ_1",
groupby="source",
top=12,
)
plt.show()
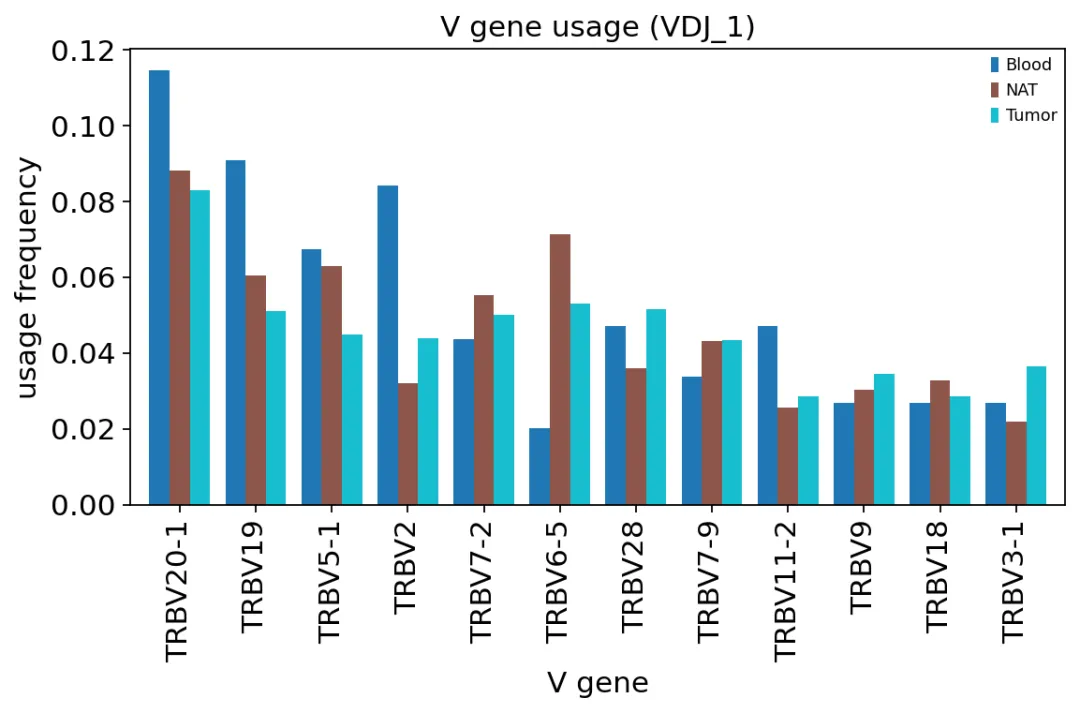 TRBV gene usage
TRBV gene usageTRBV20-1、TRBV28、TRBV9等基因占比比较高,而且Tumor、NAT和Blood之间整体差异不大。
这说明在这个数据里,组织来源之间的主要差别不是“换了一批V基因”。真正变化更明显的,是某些具体克隆在肿瘤里扩增了。
这两个概念要分开:gene usage看的是重排基因片段的整体偏好,clonal expansion看的是具体TCR克隆是否被拉起来。
10. 克隆扩增发生在哪些T细胞状态
现在进入单细胞TCR最重要的问题:克隆扩增到底对应哪些转录状态?
ct_exp = ov.airr.clonal_expansion_composition(
tcr,
groupby="cell_type",
)
ct_exp.round(3)
clonal_expansion 1 (single) 2 3 >= 4
cell_type
8.1-Teff 0.387 0.134 0.071 0.408
8.3b-Trm 0.469 0.144 0.067 0.321
8.3c-Trm 0.538 0.176 0.061 0.226
8.4-Chrom 0.652 0.087 0.043 0.217
8.2-Tem 0.611 0.152 0.065 0.172
画成堆叠柱状图:
fig, ax = plt.subplots(figsize=(7, 4.5))
ct_exp.plot(
kind="bar",
stacked=True,
ax=ax,
colormap="viridis",
width=0.82,
)
ax.set_ylabel("fraction of cells")
ax.set_xlabel("transcriptomic state")
plt.show()
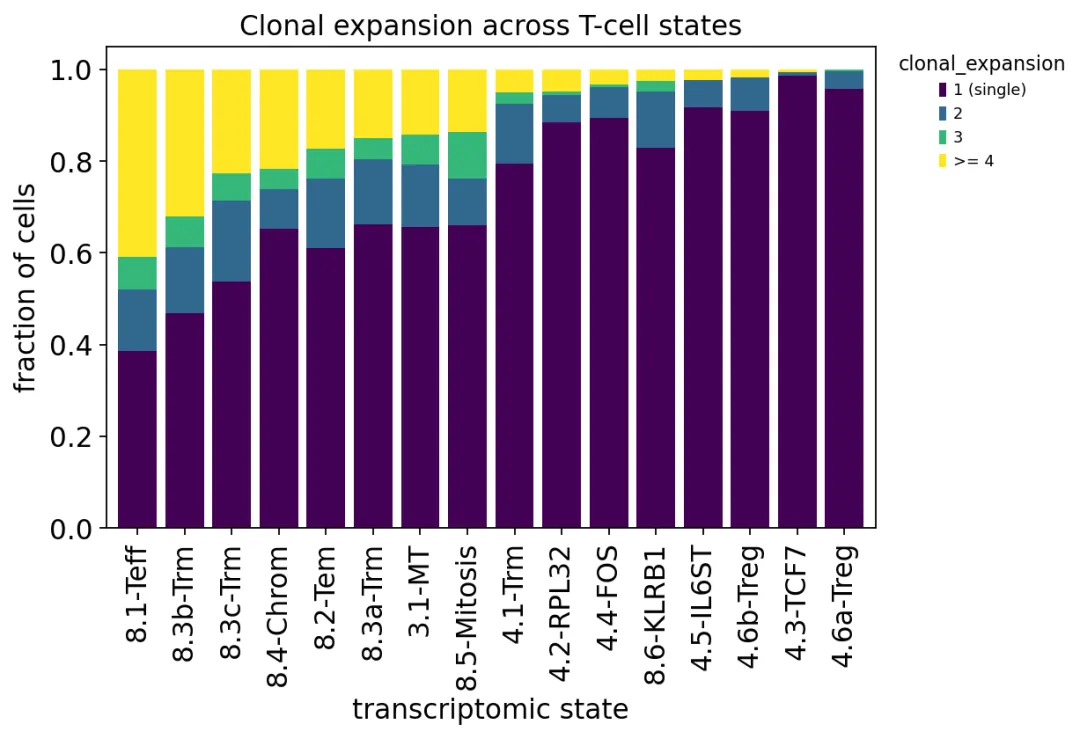 不同T细胞状态的克隆扩增比例
不同T细胞状态的克隆扩增比例我们发现,8.1-Teff、8.3b-Trm、8.3c-Trm、8.2-Tem这些CD8 effector / tissue-resident memory相关状态里,大克隆比例更高。
相反,很多CD4 naive-like或者regulatory相关状态大多是singleton。
这说明克隆扩增没有平均分布在所有T细胞状态中,它更集中在抗原经历过的CD8 T细胞状态里。
再把clone size放回原始UMAP:
adata.obs["clone_size"] = np.nan
adata.obs.loc[tcr.obs_names, "clone_size"] = tcr.obs["clone_id_size"].values
ov.pl.embedding(
adata,
basis="X_umap_orig",
color="clone_size",
frameon="small",
cmap="Reds",
show=False,
)
plt.show()
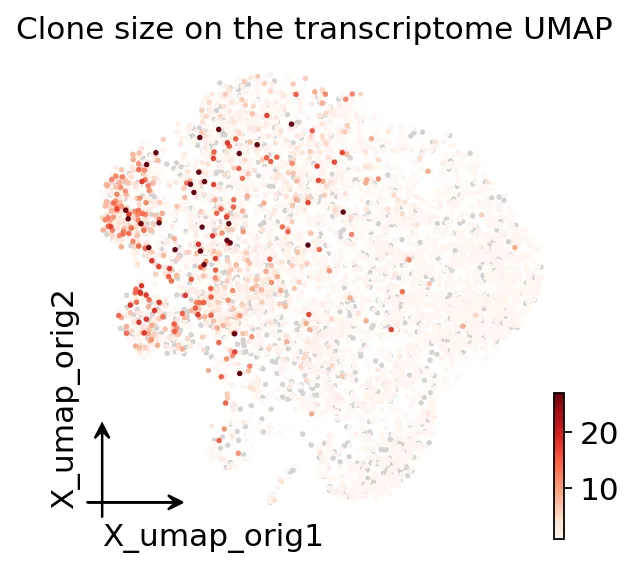 clone size投影到转录组UMAP
clone size投影到转录组UMAP可以看到,大克隆细胞没有随机散在UMAP上,而是集中在CD8 effector / Trm相关区域。
这就是为什么我们需要单细胞TCR。因为它可以把“克隆扩增”直接映射到“细胞状态”上。
11. MAIT和iNKT识别
肿瘤T细胞里还有一些先天样T细胞,比如MAIT和iNKT。
这类细胞的TCR具有比较固定的V/J组合,识别的抗原类型也和普通peptide-MHC不同。如果不提前标出来,它们可能会被误认为普通肿瘤反应性T细胞。
ov.airr.detect_invariant(tcr)
inv = tcr.obs["invariant_tcell"].value_counts()
n_innate = int(inv.get("MAIT", 0) + inv.get("iNKT", 0))
print(inv)
print(f"\ninnate-like (MAIT + iNKT): {n_innate} cells ({n_innate / tcr.n_obs:.1%} of the repertoire)")
invariant_tcell
conventional 3002
unknown 1157
MAIT 27
iNKT 2
innate-like (MAIT + iNKT): 29 cells (0.7% of the repertoire)
把MAIT/iNKT投影到UMAP上:
ax = ov.pl.embedding(
adata,
basis="X_umap_orig",
color="cell_type",
frameon="small",
title="MAIT / iNKT cells on the transcriptome",
show=False,
)
plt.show()
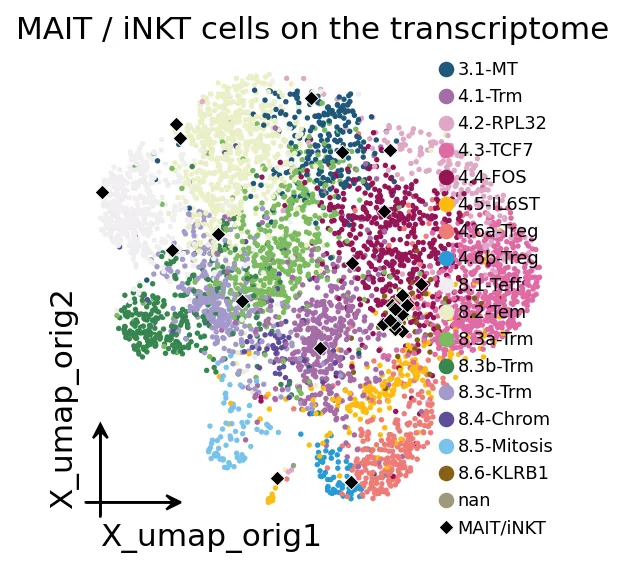 MAIT/iNKT在UMAP上的位置
MAIT/iNKT在UMAP上的位置这个数据中MAIT和iNKT数量不多,合计只有29个细胞。
但它们值得单独标记。因为这类细胞不是典型的肿瘤新抗原反应T细胞。
12. Clonotype imbalance
接下来我们想问一个更具体的问题:哪些clonotype在Tumor和Blood之间明显偏向某一侧?
imb = ov.airr.clonotype_imbalance(
tcr,
groupby="source",
case="Tumor",
control="Blood",
)
print(f"clonotypes tested : {len(imb)}")
print(f"significant (padj < 0.05): {(imb['pvalue_adj'] < 0.05).sum()}")
imb.head()
clonotypes tested : 2065
significant (padj < 0.05): 1
结果里正的log2FC代表Tumor富集,负的log2FC代表Blood富集。
# Take the 8 most blood-enriched + the 7 most tumour-enriched clones
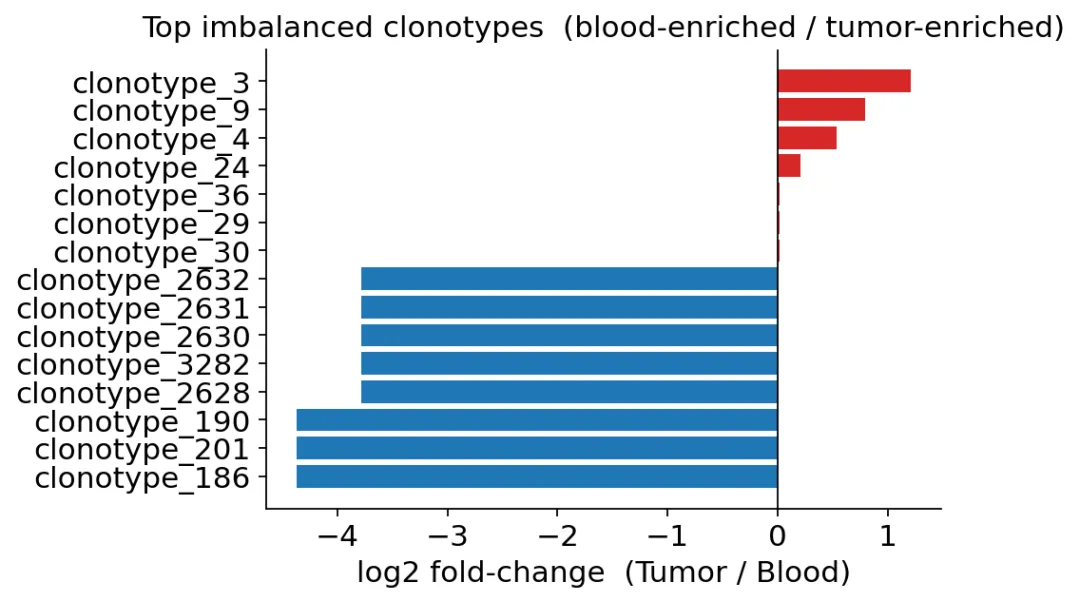 Tumor和Blood之间的clonotype imbalance
Tumor和Blood之间的clonotype imbalance这里需要注意,只有1个clonotype通过了校正后的显著性阈值。
这并不奇怪。因为Blood细胞只有300多个,单个clonotype在Blood里很稀有,统计检验会比较保守。
这张图告诉我们哪些克隆偏向Blood,哪些克隆偏向Tumor。Tumor富集的克隆可以作为候选肿瘤反应性TCR继续跟踪。
13. Clonotype转录状态一致性
同一个clonotype里的细胞,是否都处在同一个转录状态?
这个问题很有意思。因为同一个克隆扩增出来以后,它的子代细胞可能保持同一种状态,也可能分化到多个状态。
mod = ov.airr.clonotype_modularity(
tcr,
cluster_key="cell_type",
)
mod_exp = mod[mod["size"] >= 3]
print(f"clonotypes scored : {len(mod)}")
print(f"expanded (size >= 3) : {len(mod_exp)}")
print(f"mean modularity (size>=3): {mod_exp['modularity_score'].mean():.3f}")
mod_exp.head()
clonotypes scored : 3348
expanded (size >= 3) : 126
mean modularity (size>=3): 0.593
画出clone size和modularity的关系:
# Modularity vs size scatter
dominant cluster of expanded clones:
dominant_cluster
8.1-Teff 40
8.2-Tem 24
3.1-MT 16
8.3a-Trm 13
8.3b-Trm 12
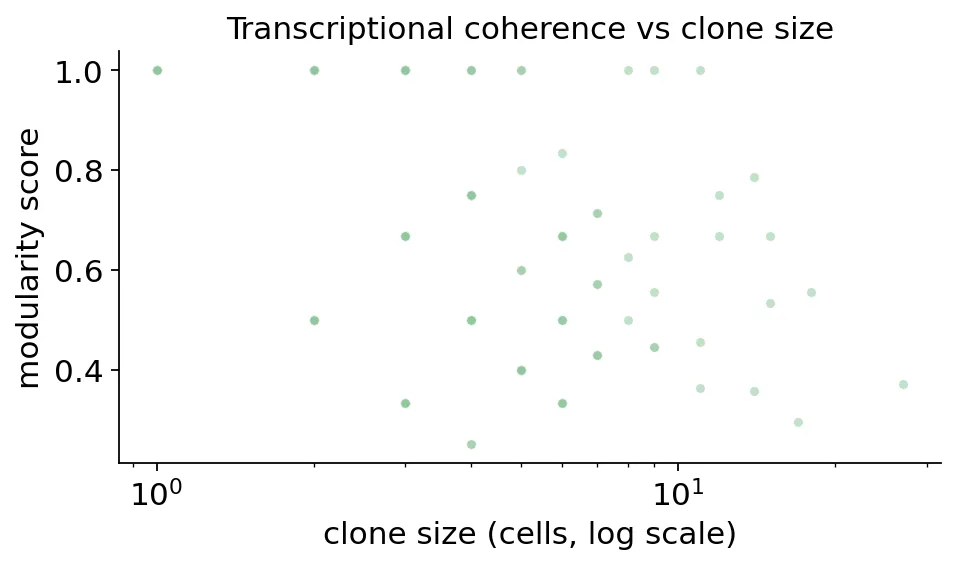 Clonotype modularity
Clonotype modularity大多数扩增克隆的dominant cluster还是集中在CD8 effector / Tem / Trm相关状态。
这说明前面看到的结果不是只有群体层面的平均现象。很多具体克隆内部也有相对一致的转录状态。
换句话说,一个肿瘤扩增克隆往往不会随机散落在所有T细胞状态里,它更偏向某个CD8效应状态。
14. CDR3 motif logo
CDR3是TCR里最可变、也最接近抗原识别的一段序列。
我们可以先看整个repertoire的CDR3β logo:
ax = ov.airr.cdr3_logo(tcr, chain="beta", kind="information")
ax.set_title("CDR3β logo — whole repertoire")
plt.show()
 全repertoire CDR3β logo
全repertoire CDR3β logo全repertoire里,CDR3两端的保守结构更明显,中间抗原接触区域则高度多样。
再看最大克隆的CDR3β logo:
top_clone = tcr.obs["clone_id"].value_counts().index[0]
sub = tcr[tcr.obs["clone_id"] == top_clone]
ax = ov.airr.cdr3_logo(sub, chain="beta", kind="information")
ax.set_title(f"CDR3β logo — {top_clone}")
plt.show()
 最大克隆的CDR3β logo
最大克隆的CDR3β logo最大克隆里,每个位置基本都是一个固定氨基酸。
这其实就是clonotype的可视化定义:很多细胞,共享同一个TCR序列。
15. VDJdb抗原注释
最后,我们把TCR序列和VDJdb数据库做匹配。
ref = ov.datasets.vdjdb_reference()
ref_trb = ref[ref["gene"] == "TRB"]
print(f"VDJdb TRB reference records : {len(ref_trb)}")
print(f"distinct epitope peptides : {ref_trb['antigen_epitope'].nunique()}")
print(f"distinct source antigens : {ref_trb['antigen_gene'].nunique()}")
print(ref_trb['antigen_species'].value_counts().head(6))
VDJdb TRB reference records : 87992
distinct epitope peptides : 1702
distinct source antigens : 285
antigen_species
HomoSapiens 35283
CMV 27544
InfluenzaA 6780
EBV 6488
SARS-CoV-2 5519
HIV-1 2812
运行注释:
ov.airr.annotate_antigen(
tcr,
reference=ref_trb,
chain="beta",
key_added="",
)
n_beta = int(ov.airr.usable_cdr3_mask(tcr, chain="beta").sum())
n_hit = int(tcr.obs["epitope"].notna().sum())
print(f"cells with a beta chain : {n_beta}")
print(f"cells matched to an epitope : {n_hit} ({n_hit/n_beta:.1%} of beta-chain cells)")
print(f"distinct epitopes matched : {tcr.obs['epitope'].nunique()}")
print()
print("top matched epitope peptides:")
print(tcr.obs["epitope"].value_counts().head(8))
cells with a beta chain : 3762
cells matched to an epitope : 108 (2.9% of beta-chain cells)
distinct epitopes matched : 30
top matched epitope peptides:
epitope
KLGGALQAK 35
NLVPMVATV 22
GILGFVFTL 6
FLRGRAYGL 4
YLQPRTFLL 3
KRWIILGLNK 3
VVTGVLVYL 3
GLCTLVAML 2
命中率只有2.9%,这是正常的。
VDJdb本身只是已知TCR-抗原关系的一小部分,而且这里用的是严格的beta链CDR3精确匹配。真正肿瘤浸润T细胞里,大部分TCR在数据库里没有完全一样的记录。
这些命中主要来自CMV、Influenza、EBV等常见病毒抗原,例如CMV的 KLGGALQAK、NLVPMVATV,Influenza的 GILGFVFTL。
在肿瘤TCR分析里,我觉得VDJdb注释最实用的地方,不是直接证明某个TCR识别肿瘤抗原。它更像是帮我们识别一部分已知病毒相关的bystander T cells。把这些已知抗病毒克隆标出来以后,剩下那些大型、patient-private、数据库未注释的肿瘤内扩增克隆,反而更值得继续追。
16. 总结
在本期教程中,我们用Wu 2020单细胞TCR + 转录组数据跑完了一套单细胞免疫组库分析流程:
- 原始数据包含5001个T细胞,来自Tumor、NAT和Blood三个来源。
- 经过TCR chain QC后,保留4188个带有可用TCR的细胞。
- 严格CDR3定义下,一共得到3348个exact clonotypes,最大clone包含27个细胞。
- 28.1%的细胞属于size >= 2的扩增克隆,肿瘤来源细胞扩增比例最高。
- Clonotype network显示很多扩增克隆同时包含Tumor和NAT细胞,提示组织间存在克隆共享。
- 患者之间的TCR overlap接近0,说明严格定义下clonotype基本是patient-private。
- TRBV gene usage在Tumor、NAT和Blood之间差异不大,主要变化来自具体克隆扩增,而不是整体V基因使用偏移。
- 克隆扩增主要集中在CD8 effector / Trm / Tem相关状态,尤其是
8.1-Teff、8.3b-Trm、8.3c-Trm。 - MAIT和iNKT合计只有29个细胞,占0.7%,可以作为先天样T细胞单独标记。
- VDJdb精确匹配得到108个细胞、30个epitopes,命中率为2.9%,主要是CMV、Influenza和EBV相关TCR。
实际上我觉得单细胞TCR分析最有价值的地方,不在于“画一个clonotype network”,而在于把TCR和转录状态放在一起看。
因为克隆扩增本身只告诉我们这个T细胞家族被拉起来了,但它不告诉我们这些细胞现在是什么状态。只有把TCR接回转录组,我们才能看到扩增克隆到底落在CD8 Teff、Trm、Treg还是naive-like状态里。
对于肿瘤免疫项目来说,这一点尤其重要。真正值得继续追的,往往是那些在肿瘤中扩增、处在CD8效应/耗竭相关状态、同时又没有被VDJdb注释成常见病毒bystander的patient-private clonotypes。
这也是我觉得OmicVerse整合 ov.airr 的原因:用户不需要在TCR包和单细胞包之间来回切换,就可以在同一个AnnData对象里完成从TCR QC到转录组联合分析的完整流程。
17. 交流群
如果你也很好奇我们omicverse生态的最新进展,想第一时间体验到新功能,欢迎加入我们的交流群~

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
