python单细胞流程-高变基因选择
- 2026-07-01 23:43:54
为什么要进行高变基因选择?
在单细胞 RNA-seq 数据中,一个表达矩阵通常会包含上万到数万个基因。但在后续分析中,并不是所有基因都同样有用。
很多基因在大部分细胞中几乎不表达,矩阵中主要是 0;也有一些基因虽然表达量很高,但在不同细胞之间变化不明显,例如某些 housekeeping genes。这些基因如果全部放入 PCA、聚类和 UMAP 等步骤中,可能会增加计算量,也可能让真正反映细胞类型差异的信号被噪音稀释。
高变基因选择(highly variable gene selection, HVG selection)的目的,是在所有基因中筛选出一部分在细胞之间变化较明显、信息量较高的基因。后续通常只使用这些高变基因进行降维、邻居图构建、聚类和可视化。
需要注意的是,高变基因不等同于 marker gene。高变基因是在不知道细胞类型之前,用于捕捉主要表达差异的一组特征基因;marker gene 则通常是在已经完成分群或注释之后,用来解释某个细胞群身份的特征基因。
高变基因的基本思想
在单细胞数据中,一个基因是否“变化大”,不能只看普通方差。因为基因表达均值和方差之间通常存在关系:表达量越高的基因,方差往往也会更高。如果直接按照方差排序,容易优先选到高表达基因,而不一定是真正区分细胞状态的基因。
因此,常见的高变基因筛选方法会先考虑基因的平均表达水平,再比较同一表达水平附近的基因变异程度。简单理解就是:
高变基因 = 在相似平均表达水平下,变化幅度明显更大的基因
在 Scanpy 中,sc.pp.highly_variable_genes 会计算每个基因的均值、离散度或标准化后的离散度,并在 adata.var 中添加高变基因标记。
本节使用已经完成的 Log(CP10k+1) 归一化数据,因此采用 flavor="seurat"。这个方法适用于已经归一化并进行 log 转换后的表达矩阵。如果使用原始 counts 数据,也可以考虑 flavor="seurat_v3",但该方法需要基于 count 数据,并且通常需要额外安装 scikit-misc。
读取归一化后的数据
import os
import scanpy as sc
adata = sc.read_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/03_pbmc_10k_normalized_log1p.h5ad")
adata
这里读入的 adata.X 已经是 Log(CP10k+1) 归一化后的表达矩阵。上一节还将完整的归一化表达矩阵保存到了 adata.raw 中,因此即使后续只保留高变基因,也仍然可以在 marker gene 可视化或细胞类型注释时访问完整基因的表达信息。
可以先查看当前对象中是否保留了原始 counts layer 和 raw 信息:
print(adata.shape)
print(adata.layers.keys())
使用 Scanpy 筛选高变基因
这里可以采用两种常见方式筛选高变基因。
第一种方式是固定选择 2000 个高变基因,这是单细胞分析中比较常见的默认数量。对于 PBMC 这类常规数据集,2000 个高变基因通常足够捕捉主要细胞类型差异,也能降低后续降维和聚类时的噪音。
sc.pp.highly_variable_genes(
adata,
n_top_genes=2000,
flavor="seurat"
)
第二种方式是根据平均表达量和标准化离散度设置阈值:
sc.pp.highly_variable_genes(
adata,
min_mean=0.0125,
max_mean=3,
min_disp=0.5,
flavor="seurat"
)
这几个参数的含义是:
min_mean=0.0125:过滤掉平均表达量过低的基因。这类基因在大部分细胞中几乎不表达,容易受到 dropout 和稀疏噪音影响。max_mean=3:过滤掉平均表达量过高的基因。这类基因可能是普遍高表达基因,不一定能有效区分不同细胞群。min_disp=0.5:保留标准化离散度较高的基因,也就是在相似平均表达水平下变化更明显的基因。
这两种方式的区别在于:n_top_genes=2000 是固定选择前 2000 个高变基因;而 min_mean、max_mean 和 min_disp 是按阈值筛选,最后得到的高变基因数量不固定,具体取决于数据本身。
需要注意的是,n_top_genes 和这些阈值参数通常二选一使用。如果连续运行两段代码,后一次运行会覆盖前一次写入 adata.var["highly_variable"] 的结果。实际分析中选择其中一种方式即可。
执行完成后,Scanpy 会在 adata.var 中添加几列新的信息:
highly_variable:是否被选为高变基因,值为 True 或 False。means:每个基因的平均表达水平。dispersions:每个基因的离散度。dispersions_norm:经过表达水平校正后的标准化离散度。
可以查看这些结果:
adata.var[["highly_variable", "means", "dispersions", "dispersions_norm"]].head()
统计被选中的高变基因数量:
adata.var["highly_variable"].sum()
np.int64(2000)
如果使用固定数量法,这里通常会得到 np.int64(2000);如果使用阈值法,结果则不一定是 2000,而是由 min_mean、max_mean 和 min_disp 共同决定。
如果想查看标准化离散度最高的一些高变基因,可以按照 dispersions_norm 排序:
adata.var.loc[
adata.var["highly_variable"],
["means", "dispersions", "dispersions_norm"]
].sort_values("dispersions_norm", ascending=False).head(20)
查看高变基因结果
Scanpy 提供了专门的函数来可视化高变基因筛选结果:
sc.pl.highly_variable_genes(adata)
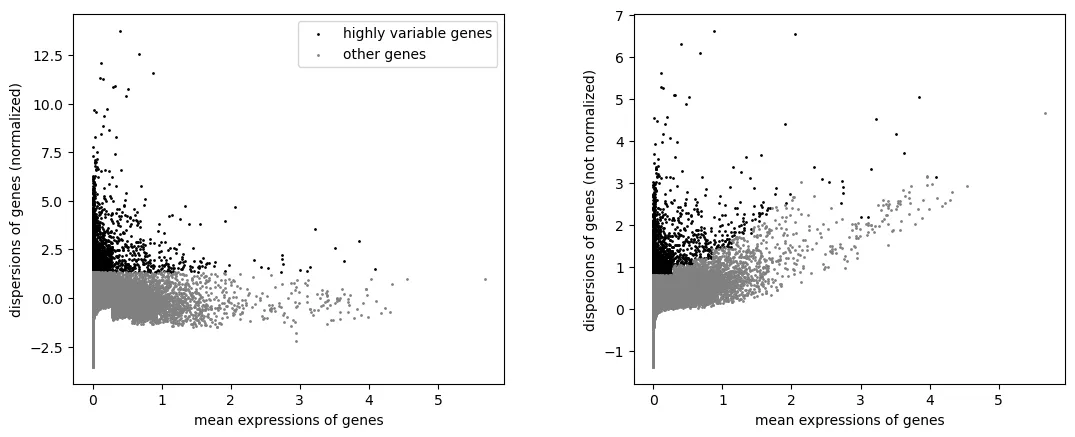
左图展示的是标准化后的离散度。横轴是基因的平均表达量,纵轴是经过表达量校正后的离散度。高变基因通常位于纵轴较高的位置,表示这些基因在相似平均表达水平下,比其他基因变化更明显。 右图展示的是未标准化的离散度。可以看到,平均表达量较高的基因往往天然具有更高的原始离散度,因此不能只根据右图直接判断高变基因。
因此,判断这张图时主要看左图:被选中的黑色点应该是在不同平均表达水平下标准化离散度较高的一批基因,而不是简单集中在最高表达区域。
还可以分别查看高变基因和非高变基因的数量:
adata.var["highly_variable"].value_counts()
如果后续想检查某些已知 marker 是否被选中,可以直接查询:
genes = ["MS4A1", "CD3D", "LYZ", "NKG7"]
adata.var.loc[
adata.var_names.intersection(genes),
["highly_variable", "means", "dispersions_norm"]
]
需要注意的是,某个 marker gene 没有被选为高变基因,并不代表它不重要。高变基因主要服务于降维和聚类;marker gene 展示、细胞类型注释和差异表达分析仍然可以使用完整基因矩阵,尤其是前面保存到 adata.raw 中的表达信息。
是否只保留高变基因?
完成高变基因筛选后,有两种常见做法。
第一种做法是不立即删除其他基因,只是在 adata.var["highly_variable"] 中保留标记。这样对象仍然保留完整基因矩阵,后续某些步骤可以根据需要指定是否只使用高变基因。
第二种做法是直接将表达矩阵 subset 到高变基因,只保留高变基因用于后续 PCA、聚类和 UMAP:
adata = adata[:, adata.var["highly_variable"]].copy()
adata
在标准 Scanpy 流程中,通常会在高变基因筛选后只保留高变基因,然后继续进行 scaling、PCA 和聚类分析。这样可以减少无信息基因带来的噪音,也能加快后续计算。
但是,在执行 subset 前需要确认已经保存了完整表达矩阵。上一节中已经设置:
adata.raw = adata.copy()
因此,即使当前 adata.X 只剩高变基因,后续仍然可以通过 adata.raw 访问所有基因的 Log(CP10k+1) 表达值。
保存
保存高变基因筛选后的对象:
adata.write_h5ad("/home/data/t090639/project/pbmc_10k/data/processed/04_pbmc_10k_hvg.h5ad")
检查文件是否保存成功:
os.path.exists("/home/data/t090639/project/pbmc_10k/data/processed/04_pbmc_10k_hvg.h5ad")
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 关于python方面的一些知识!
- Codex 有了 Python SDK:AI 编程要从工具栏走进你的产品了
- 瑞芯微Linux课程送板子~
- 2026年星芒算法挑战赛(Python小高组)—预选赛—21~30题解题思路
- Python字符串学习蓝图
- linux kernel的virtual kernel memory layout介绍(aarch64)
- 为什么需要系统学习Linux底层驱动
- 疑似巴基斯坦关联的APT36组织针对Linux平台展开攻击活动
- 名师讲堂|使用 Python 提取城市共同边界、边界乡镇识别及距离计算
- 熬三天搞不定回测框架快崩溃?这个Python神器几行代码搞定策略回测与参数优化
