多组学揭示胆固醇驱动的巨噬细胞代谢重编程及其在慢性阻塞性肺疾病中的炎症作用
题目:Multi-omics reveals cholesterol-driven macrophage metabolic reprogramming and inflammation in chronic obstructive pulmonary disease
摘要
背景: 慢性阻塞性肺疾病(COPD)是一种进行性炎症性疾病,其全球发病率和死亡率持续上升。越来越多的证据表明,全身代谢改变,尤其是血脂异常,参与了 COPD 的发病过程。然而,脂质失调与肺部炎症及组织损伤之间的机制尚未明确。方法: 对健康人群及Ⅲ–Ⅳ期 COPD 患者的血浆样本进行非靶向代谢组学分析,以识别与疾病相关的代谢改变。建立高胆固醇饮食(HCD)小鼠模型,并联合或不联合长期香烟烟雾暴露,用以评估全身胆固醇升高对肺结构和炎症的影响。采用 THP-1 来源及骨髓来源的巨噬细胞,评估胆固醇诱导的线粒体功能障碍、ROS 产生及其下游炎症信号通路。通过转录组学分析筛选关键分子介导因素。结果: 血浆代谢组学显示,COPD 患者脂质通路显著紊乱,且胆固醇水平升高与肺功能呈负相关。在体内实验中,HCD 饮食诱导肺部炎症,并进一步加重香烟烟雾所致的肺泡破坏。在巨噬细胞中,胆固醇负荷与香烟烟雾提取物联合处理破坏线粒体完整性,降低呼吸能力,并增加 ROS 产生。过量 ROS 上调 PPIA,进而激活 NF-κB 信号并增强 IL-1β 分泌。沉默 PPIA 或抑制 ROS 可减弱 NF-κB 激活及细胞因子释放。与此一致,HCD 饮食并暴露于香烟烟雾的小鼠肺组织中 PPIA 表达和 NF-κB 磷酸化水平升高,且 COPD 患者支气管肺泡灌洗液中 PPIA 水平升高。结论: 本研究揭示了一条由胆固醇驱动的代谢-炎症通路,其中巨噬细胞中线粒体功能障碍及 ROS 依赖性的 PPIA–NF-κB 轴激活促进了 COPD 的持续性肺部炎症。这些发现阐明了全身胆固醇失调与 COPD 进展之间的机制联系,并提示胆固醇代谢及线粒体稳态可能成为潜在治疗靶点。关键词: 慢性阻塞性肺疾病;代谢组学;胆固醇代谢;线粒体功能障碍;活性氧;脯氨酰顺反异构酶 A;肺部炎症结果
COPD 患者血浆胆固醇水平升高
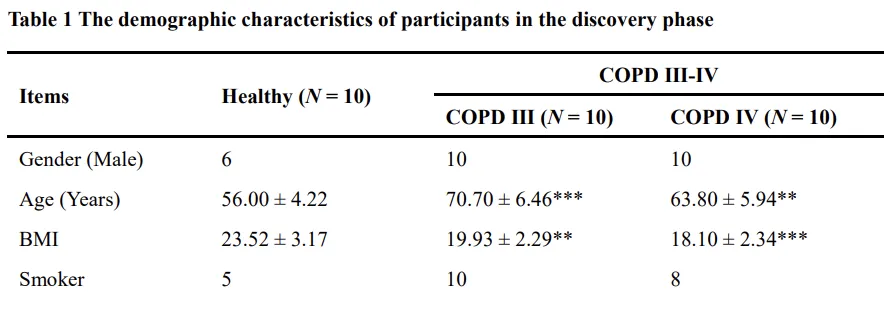
COPD 患者已知具有较高的心血管共病风险,提示可能存在全身性代谢改变[32,33]。为表征这些代谢变化,我们对健康个体和Ⅲ–Ⅳ期 COPD 患者进行了血浆代谢组学分析。研究对象的人口学特征见表1及附加文件1:表S3。 非靶向代谢组学分析在发现阶段显示两组样本在代谢谱上明显分离,提示存在与疾病相关的代谢改变(图1A)。监督式 OPLS-DA 模型进一步证实了这种分离(图1B)。多变量与单变量联合分析共鉴定出387个差异峰,其中 COPD 血浆中有199个代谢物显著上调、188个显著下调(图1C)。 基于获得的色谱和质谱信息并与公共数据库及内标进行匹配,共成功注释59种代谢物,其中大多数为脂质或类脂分子(附加文件1:表S4)。通路富集分析显示共有14条代谢通路显著扰动,主要涉及甘油磷脂代谢、亚油酸代谢以及初级胆汁酸生物合成等(图1D)。进一步的代谢物集合富集分析突出显示9条显著富集的通路,其中胆汁酸生物合成和甜菜碱代谢紊乱最为明显(图1E)。鉴于胆固醇在胆汁酸代谢中的核心作用,我们进一步分析了其与肺功能之间的关系。相关性分析显示,血浆胆固醇水平与 FEV₁/FVC 比值及 FEV₁ 预测值百分比呈显著负相关(相关系数 >0.5,P < 0.01;图1F–G)。直接定量结果证实,随着疾病严重程度的进展,血浆胆固醇水平逐步升高,这一发现在主要研究队列及一个独立的 COPD 队列中均得到验证(图1H–I)。综上所述,这些结果表明,全身性胆固醇失调是 COPD 中突出的代谢改变之一,并可能通过与肺功能受损的相关性促进疾病进展。高胆固醇饮食加重香烟烟雾暴露小鼠的肺泡破坏和肺部炎症
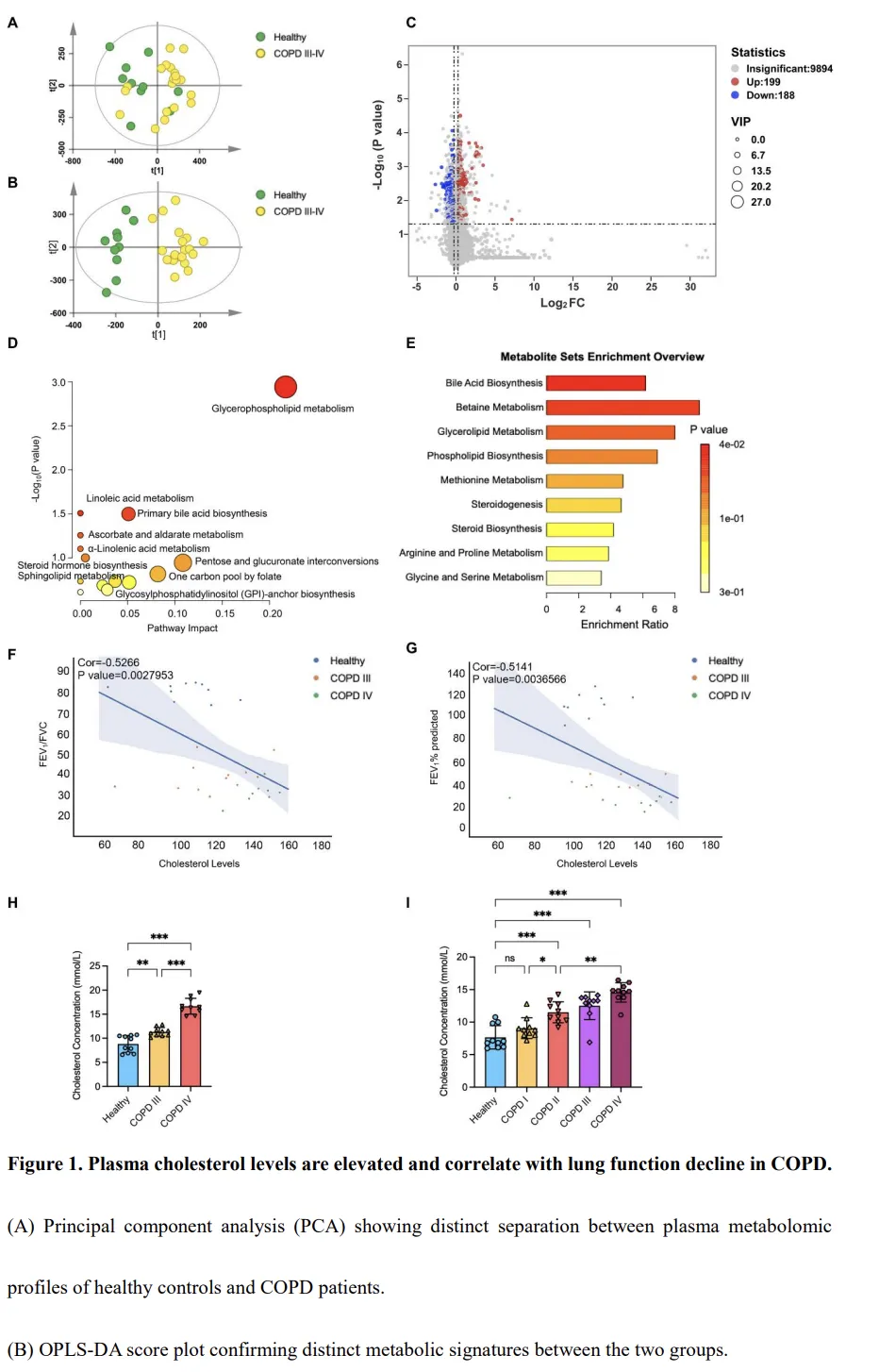
为进一步探讨膳食胆固醇对肺部病理改变的作用,我们建立了为期 6 个月的高胆固醇饮食(HCD)小鼠模型,并联合或不联合持续香烟烟雾(CS)暴露(图2A)。与正常饮食小鼠相比,HCD 饮食在空气暴露组和 CS 暴露组中均未对体重产生显著影响;然而,无论饮食条件如何,CS 暴露本身均导致体重持续下降(图2B)。如预期所示,与正常饮食对照相比,HCD 饮食小鼠组织中的胆固醇水平显著升高,且 CS 暴露进一步增强了肺组织中的胆固醇积累(图2C)。通过 Oil Red O 染色对脂滴沉积进行可视化分析。CS 暴露显著增加了脂质积累,其中 CS 与 HCD 联合处理的小鼠脂质水平最高(图2D)。这些脂滴主要定位于巨噬细胞内,而在 CS 和 HCD 条件下,肺组织中巨噬细胞的数量亦明显增加(图2D)。肺功能检测显示,与正常饮食的 CS 暴露小鼠(CS-Normal 组)相比,同时接受 CS 和 HCD 处理的小鼠(CS-Chol 组)的 Cchord 和 FVC 显著升高(P < 0.05,图2E),提示肺功能受损。组织病理学分析进一步表明,CS-Chol 小鼠出现了广泛的肺泡破坏和气道上皮增厚,其损伤程度明显超过 CS-Normal 小鼠(P < 0.001,图2F–G)。炎症分析显示,CS-Chol 组支气管肺泡灌洗液(BALF)中巨噬细胞比例升高,且肺组织中 IL-1β 水平显著增加(图2J–K),而 TNF-α 水平未见明显变化(图2L)。相反,抗炎细胞因子 IL-10 在 CS-Chol 小鼠中显著降低(P < 0.05,图2M)。综上所述,这些结果表明,膳食胆固醇通过促进结构性破坏、增强炎症细胞浸润并扰乱细胞因子稳态,加重了香烟烟雾诱导的肺损伤。这些发现支持胆固醇积累可加速 COPD 进展的结论。香烟烟雾暴露破坏肝脏脂质代谢及胆固醇稳态
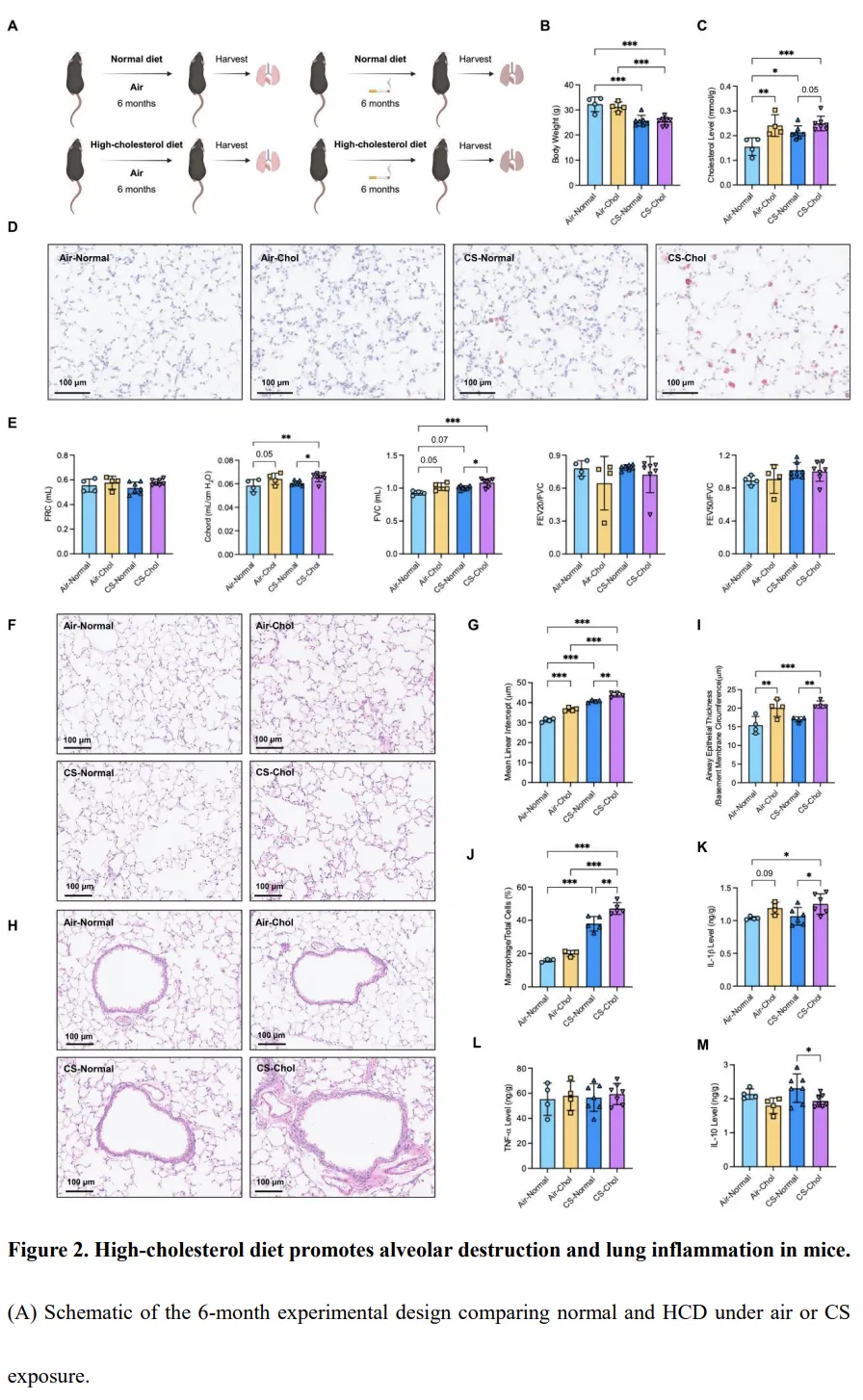
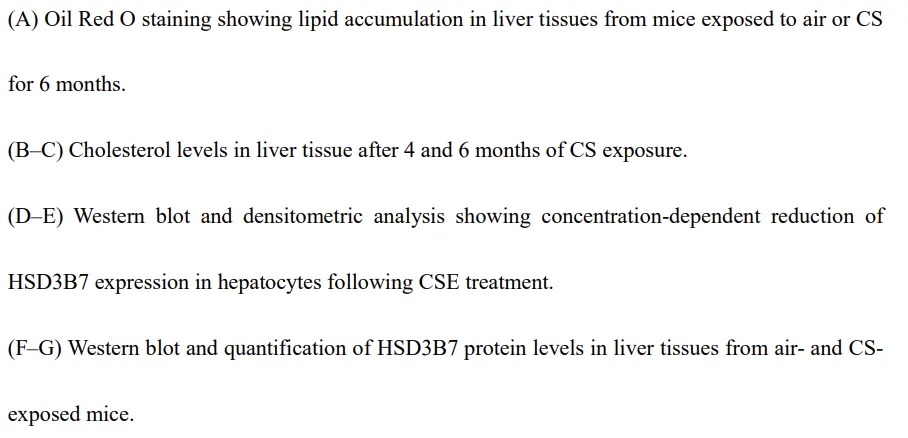
肝脏是胆固醇合成、分解及整体脂质稳态调控的核心器官。在香烟烟雾(CS)暴露的小鼠中,可观察到脂滴在肝细胞核周围明显积聚,而正常空气条件下饲养的小鼠则未见该现象(图3A)。鉴于大部分胆固醇在肝细胞中被转化为初级胆汁酸,我们对暴露 4 个月或 6 个月后采集的肝组织进行了靶向代谢组学分析。结果显示,肝脏胆固醇显著积累,且其水平随暴露时间延长而逐步升高(P < 0.05,图3B–C;附加文件1:表S5)。与此同时,多种胆汁酸亚型的肝脏水平在 CS 暴露后明显下降,包括 24(S)-羟基胆固醇、23-去甲胆酸、熊去氧胆酸以及牛磺熊去氧胆酸(P < 0.05,附加文件1:表S5)。为明确胆汁酸合成受损的机制,我们检测了 HSD3B7 的表达情况。HSD3B7 是将胆固醇转化为初级胆汁酸的关键限速酶。在 CSE 处理的肝细胞中,HSD3B7 表达呈剂量依赖性显著下调(图3D–E)。与体外结果一致,CS 暴露小鼠的肝组织中 HSD3B7 表达同样降低(图3F–G)。这些结果表明,长期 CS 暴露通过抑制 HSD3B7 表达损害肝脏胆固醇分解代谢,导致肝脏胆固醇积累。该肝脏代谢紊乱可能参与了 COPD 发病过程中所观察到的全身及肺部胆固醇失衡。胆固醇加重香烟烟雾诱导的巨噬细胞线粒体功能障碍及代谢重编程
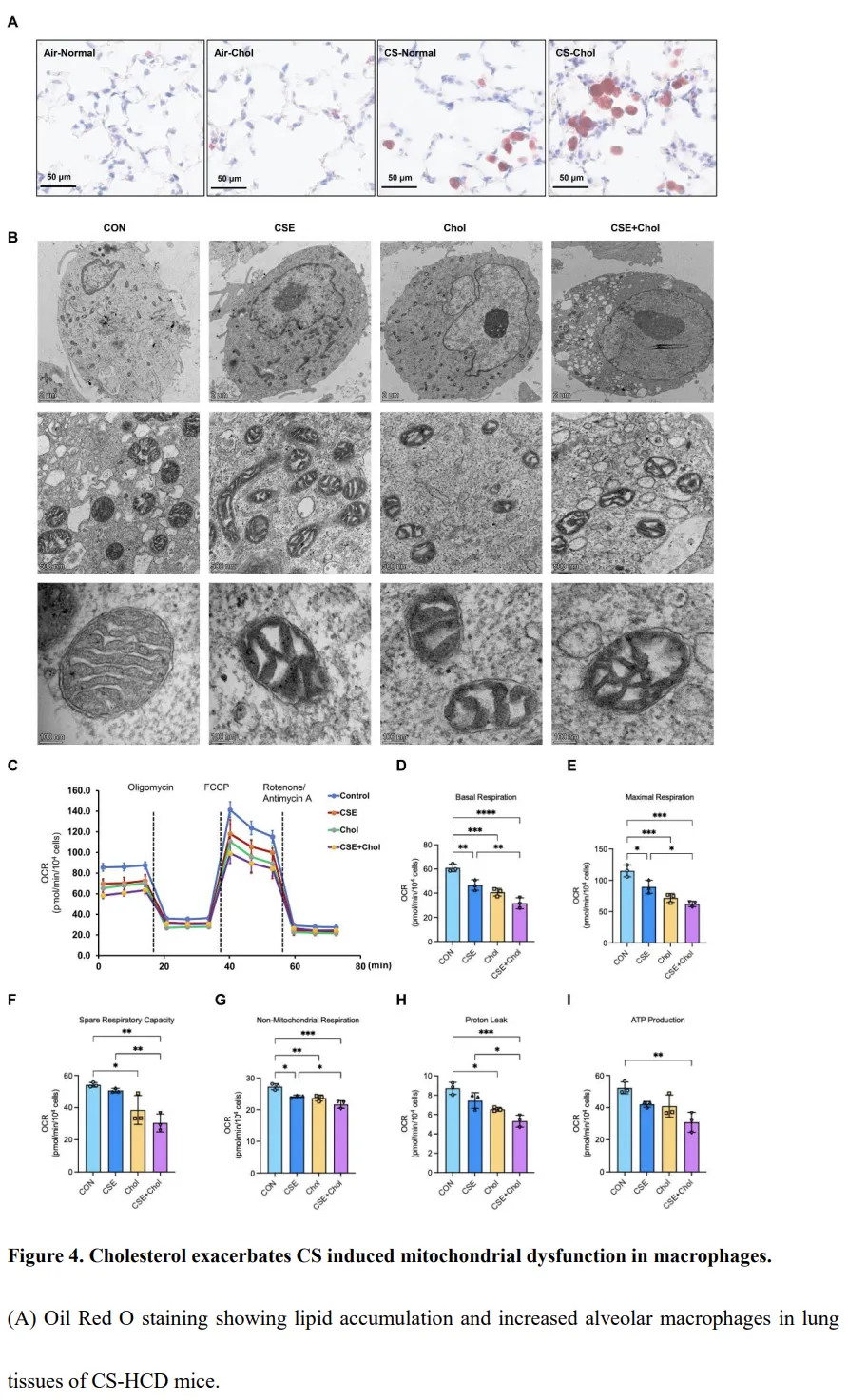
Oil Red O 染色显示,香烟烟雾(CS)暴露小鼠的肺泡巨噬细胞中脂质明显积累(图4A)。与正常饮食的 CS 暴露小鼠(CS-Normal)相比,同时接受 CS 和高胆固醇饮食(CS-Chol)的小鼠,其肺泡腔内巨噬细胞的数量和体积均明显增加(图2D、2J 和 4A),提示胆固醇驱动的巨噬细胞肥大及脂质负荷增加。为进一步评估巨噬细胞的超微结构,我们在透射电镜下观察了经香烟烟雾提取物(CSE)、胆固醇或二者联合处理后的 THP-1 来源巨噬细胞。处理后的细胞线粒体出现明显肿胀、分布不规则及嵴结构破坏,其中以 CSE 联合胆固醇处理组(CSE+Chol)最为显著,提示严重的线粒体损伤(图4B)。与上述形态学异常一致,氧消耗率(OCR)分析显示,CSE 或胆固醇处理均显著降低基础呼吸和最大呼吸水平,而联合处理时抑制作用最为明显(图4C–E)。同样,与对照组相比,胆固醇处理或 CSE+胆固醇联合处理的细胞,其备用呼吸能力和非线粒体呼吸均显著下降(图4F–G)。质子泄漏在胆固醇组及 CSE+胆固醇组均有所降低(图4H),而 ATP 生成的显著下降仅见于 CSE+胆固醇组(图4I)。总体而言,这些结果表明,CS 暴露破坏了肺泡巨噬细胞的脂质稳态,而额外的胆固醇负荷进一步加重了线粒体功能障碍。CSE 与胆固醇的协同作用导致线粒体生物能量学受损及代谢重编程,从而促成 COPD 中巨噬细胞功能异常。高胆固醇饮食上调香烟烟雾暴露小鼠肺组织中 PPIA 的表达
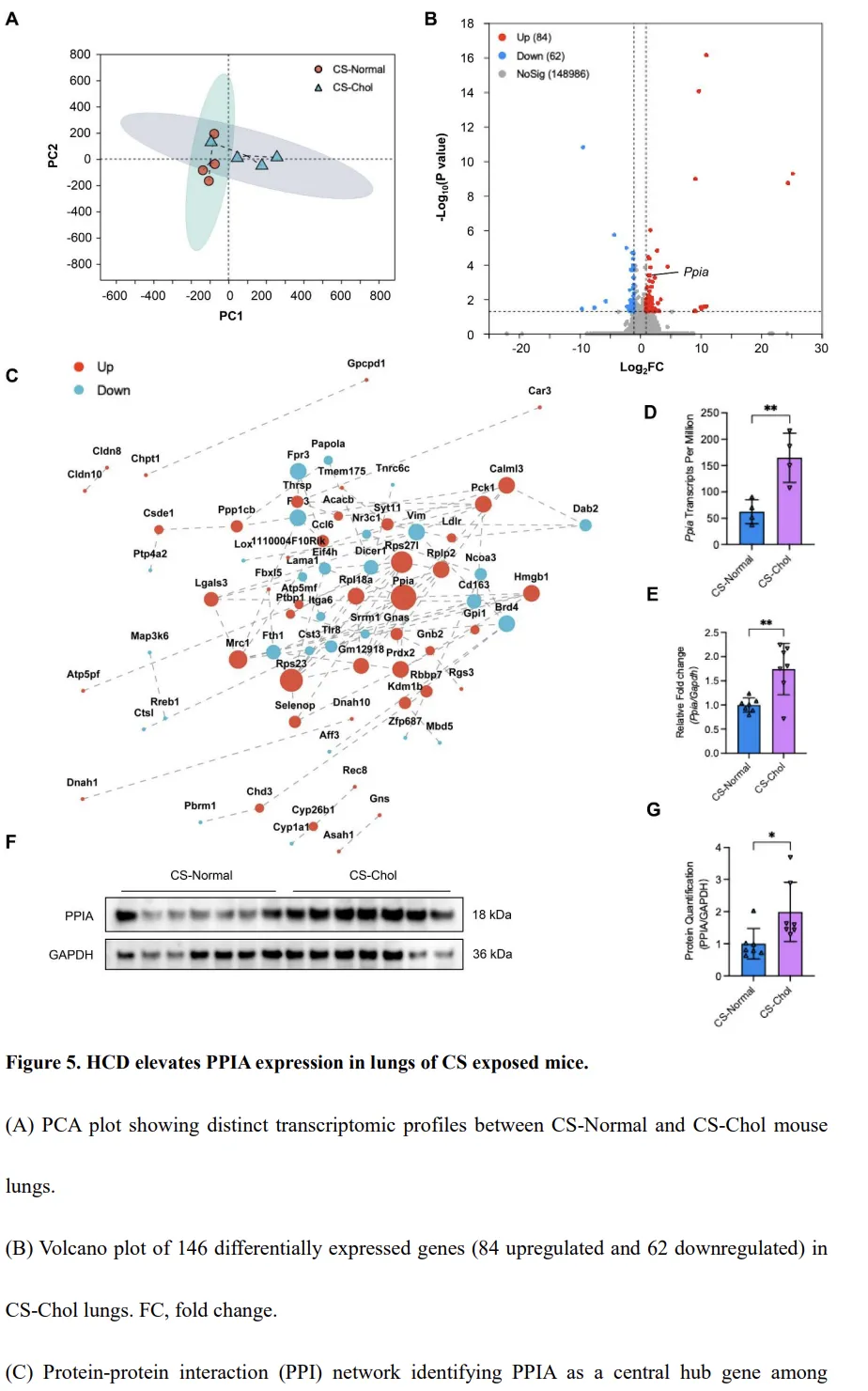
为探讨高胆固醇饮食(HCD)影响 COPD 进展的分子机制,我们对 CS-Normal 与 CS-Chol 小鼠的肺组织进行了 RNA 测序分析。主成分分析(PCA)结果显示,两组样本在转录水平上明显分离,提示膳食胆固醇相关的转录特征存在显著差异(图5A)。在 CS-Normal 与 CS-Chol 小鼠之间共鉴定出 146 个差异表达基因(DEGs),其中 84 个基因上调,62 个基因下调(图5B)。对这些 DEGs 进行蛋白–蛋白相互作用网络分析发现,脯氨酰顺反异构酶 A(Ppia)位于网络中心节点,与多种胆固醇应答相关基因存在广泛连接,提示其可能在胆固醇加重的 COPD 病理过程中发挥调控作用(图5C)。与 CS-Normal 对照相比,CS-Chol 小鼠肺组织中 Ppia 的转录水平均显著升高(图5D),该结果进一步通过 qPCR 得到验证(图5E)。Western blot 分析同样证实,CS-Chol 小鼠肺组织中 PPIA 蛋白表达显著增加(图5F–G)。这些结果表明,膳食胆固醇在香烟烟雾暴露的肺组织中,于转录及蛋白水平双重上调 PPIA 的表达。胆固醇诱导的 ROS 产生上调 PPIA 并激活巨噬细胞中的 NF-κB 信号通路
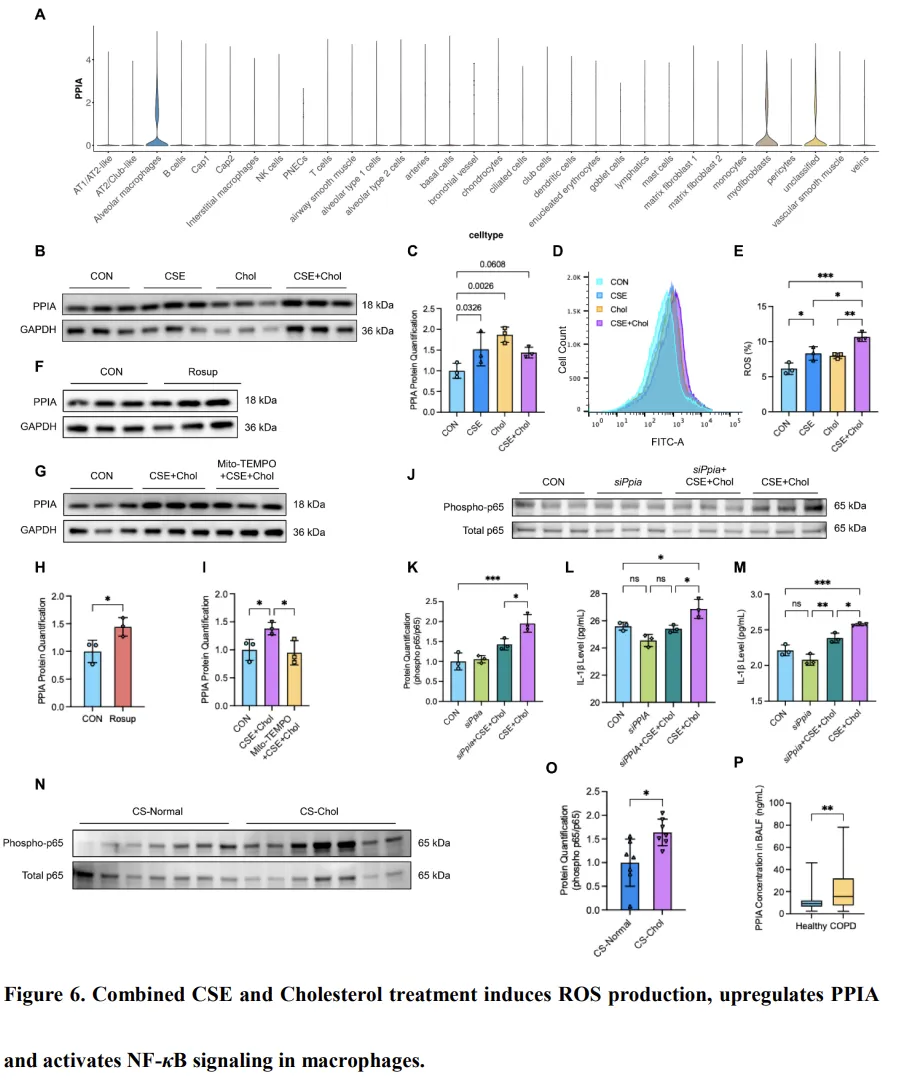
来自健康人肺组织的单细胞 RNA 测序(scRNA-seq)数据显示,PPIA 主要表达于肺泡巨噬细胞和肌成纤维细胞中(图6A)。结合我们此前的结果显示,脂质主要积聚于 CS-Chol 小鼠的肺泡巨噬细胞中(图2D 和 4A),因此我们进一步检测了 THP-1 来源巨噬细胞中 PPIA 的表达情况。结果显示,CSE 或胆固醇处理后 PPIA 表达显著升高,且在联合处理(CSE+Chol)条件下呈进一步上升趋势(图6B–C)。鉴于 CSE+Chol 处理可诱导线粒体功能障碍并增加质子泄漏,我们进一步评估了 THP-1 来源巨噬细胞中的 ROS 生成情况。如预期所示,与未处理对照相比,CSE+Chol 处理显著增加了细胞内 ROS 水平(P < 0.001,图6D–E)。此外,ROS 激动剂 Rosup 处理可增强 PPIA 的表达,而在骨髓来源巨噬细胞(BMDMs)中,ROS 抑制可有效抑制 CSE+Chol 诱导的 PPIA 上调(P < 0.05,图6F–I)。这些结果表明,胆固醇诱导的 ROS 产生可促进巨噬细胞中 PPIA 的表达。鉴于已有研究表明 PPIA 参与 NF-κB 信号通路的激活[34],我们进一步在胆固醇和 CSE 刺激条件下评估了该信号轴。在体外实验中,CSE+Chol 处理显著增强了 BMDMs 中 p65 的磷酸化水平,而 Ppia 敲低则明显减弱了 p65 的激活(P < 0.001,图6J–K)。相应地,在暴露于 CSE+Chol 的 THP-1 来源巨噬细胞和 BMDMs 中,沉默 PPIA 显著降低了 IL-1β 的分泌(P < 0.05,图6L–M)。与体外结果一致,CS-Chol 小鼠肺组织中磷酸化 p65 水平亦显著高于 CS-Normal 对照组(图6N–O)。最后,对研究队列支气管肺泡灌洗液(BALF)的分析显示,与健康对照相比,COPD 患者的 PPIA 水平显著升高,进一步支持了上述发现的临床相关性(P < 0.01,图6P)。