Python-GPU分子动力学模拟和分析流程-全网唯一出版
方法一:OpenMM(推荐用于快速测试)
安装依赖
pip install -r requirements.txt --break-system-packages
conda install -c conda-forge openmm pdbfixer ambertools
运行模拟
运行分析
方法二:GROMACS(推荐用于生产运行)
前置要求
GROMACS (已安装GPU版本)
ACPYPE (配体参数化)
pip install acpype --break-system-packages
运行模拟
python md_simulation_gmx.py
运行分析
# GROMACS内置分析
python md_analysis_gmx.py
# 或使用MDTraj分析(需要先转换轨迹)
cd gmx_output
echo"1" | gmx trjconv -s md.tpr -f md_noPBC.xtc -o trajectory.pdb -dt100
cd ..
# 修改md_analysis.py中的路径后运行
python md_analysis.py
输出文件
模拟文件
分析结果(analysis/ 或 gmx_analysis/)
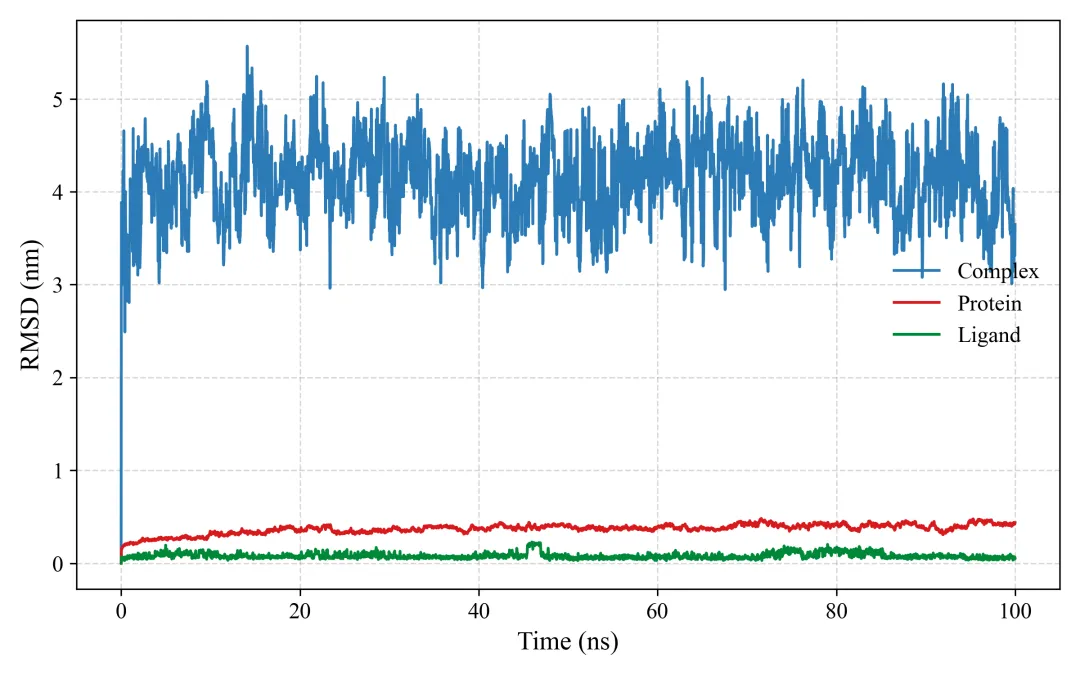
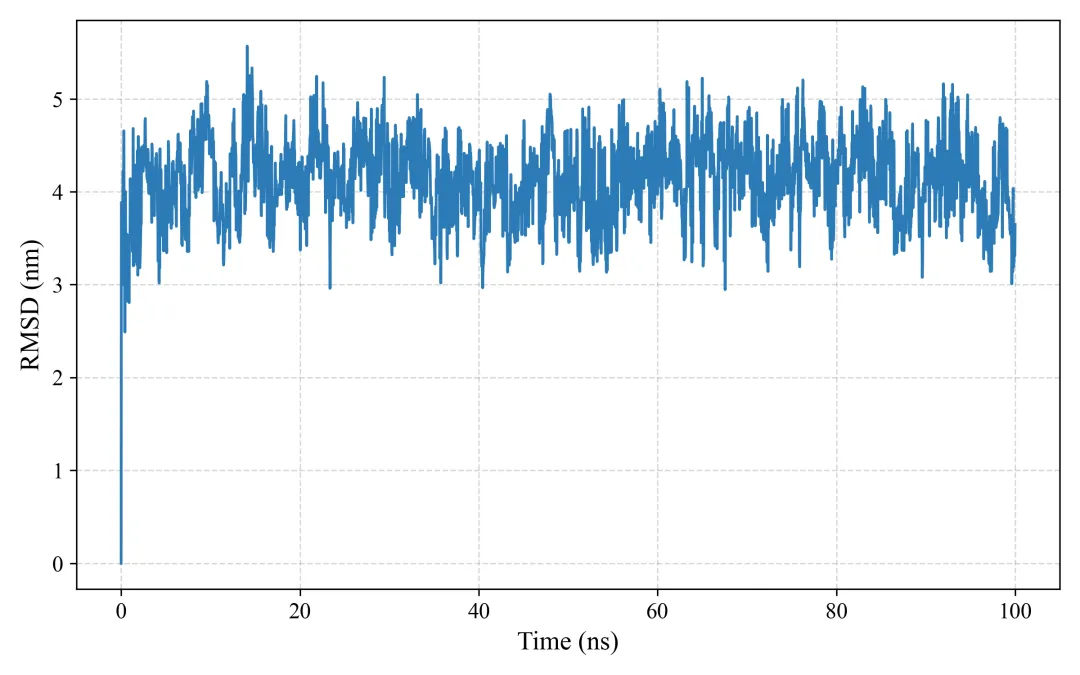
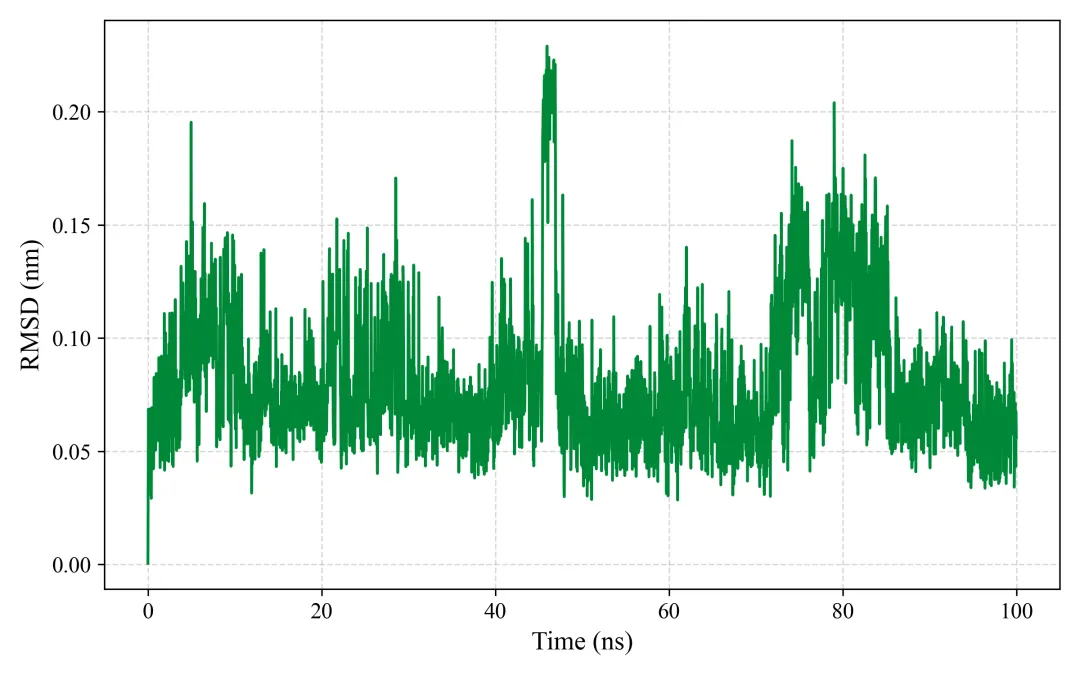
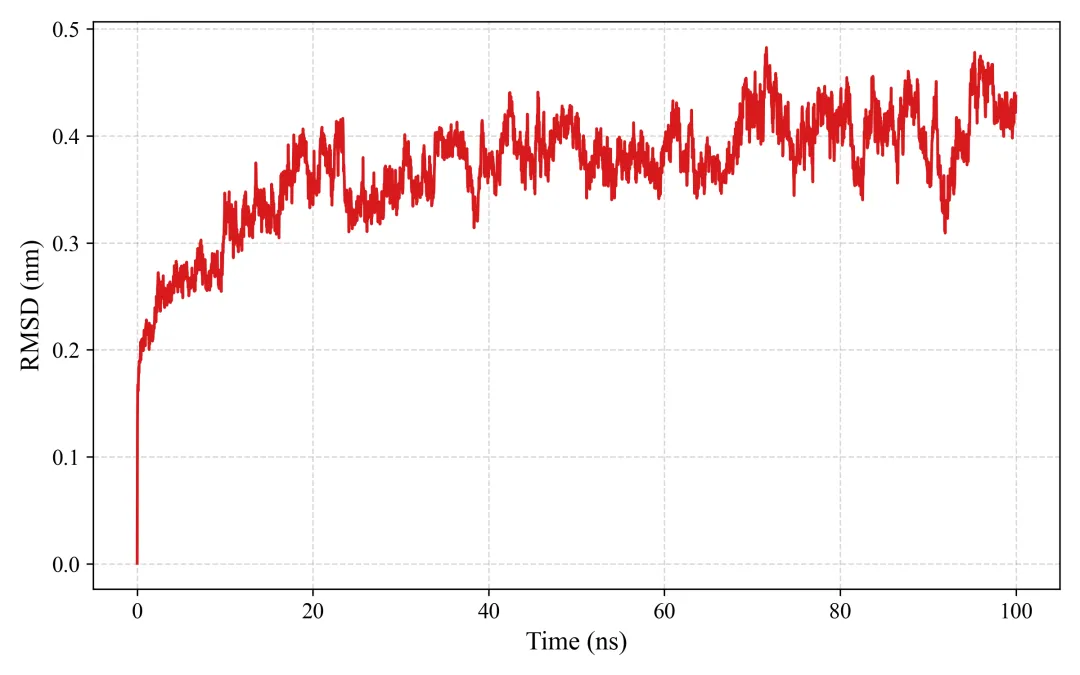
rmsd.png/pdf: RMSD图
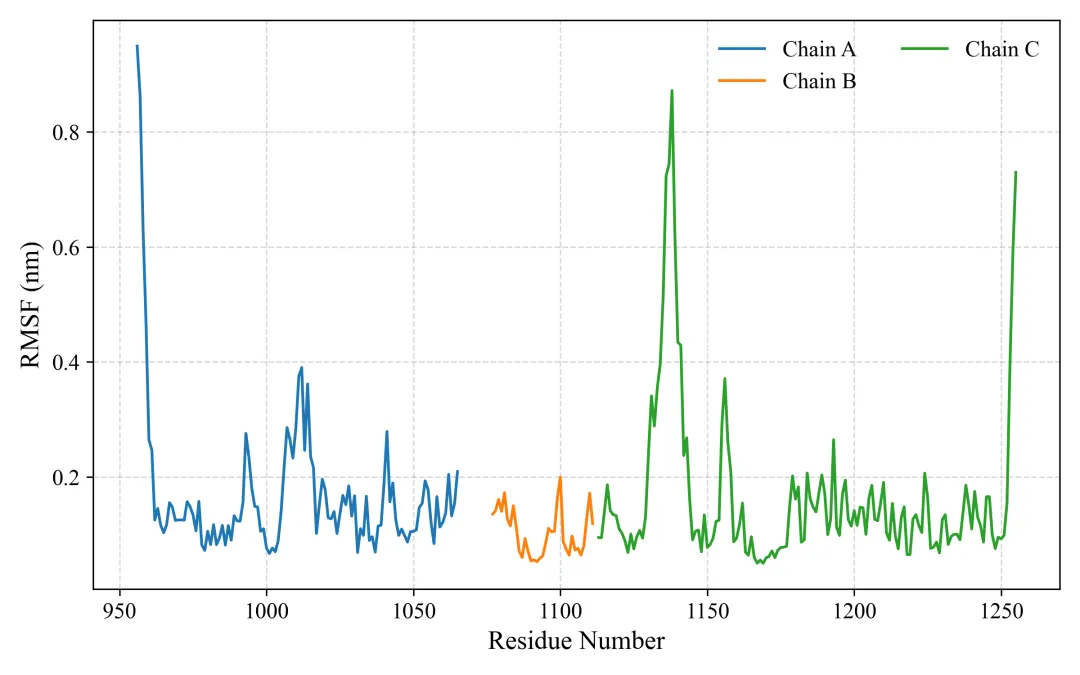
rmsf.png/pdf: RMSF图
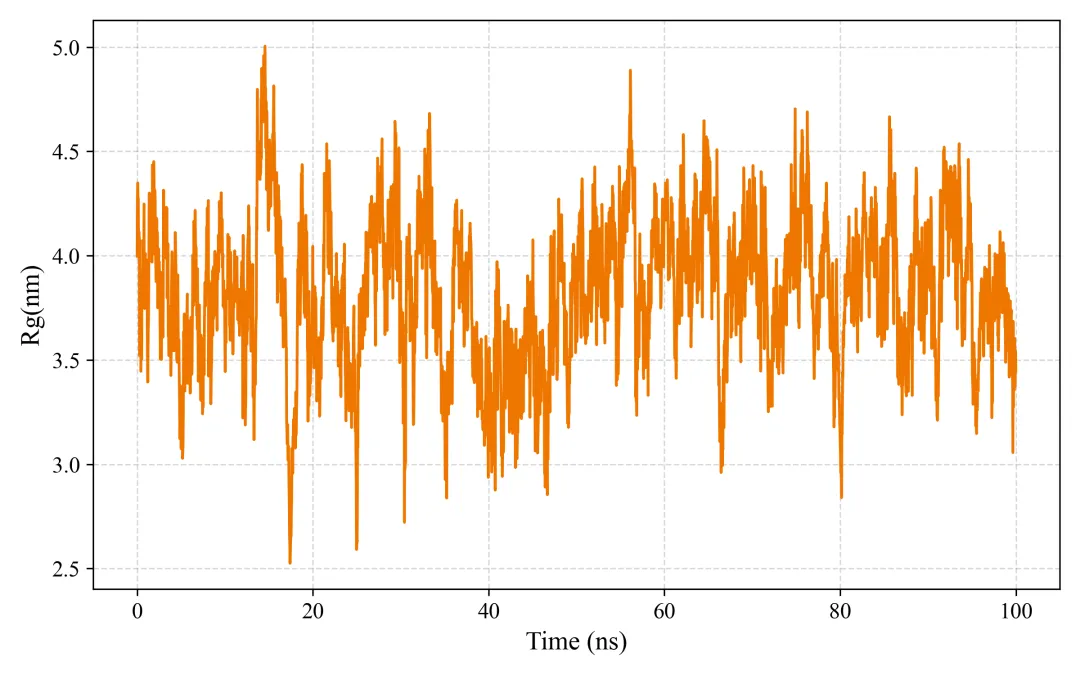
rg.png/pdf: 回旋半径
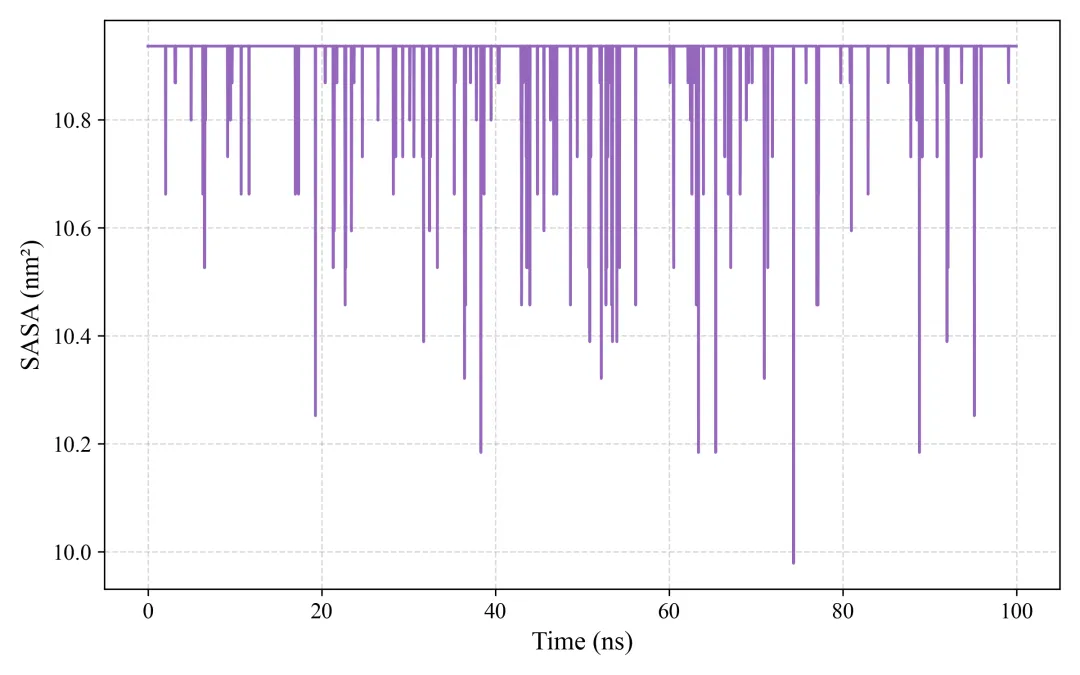
sasa.png/pdf: 溶剂可及表面积
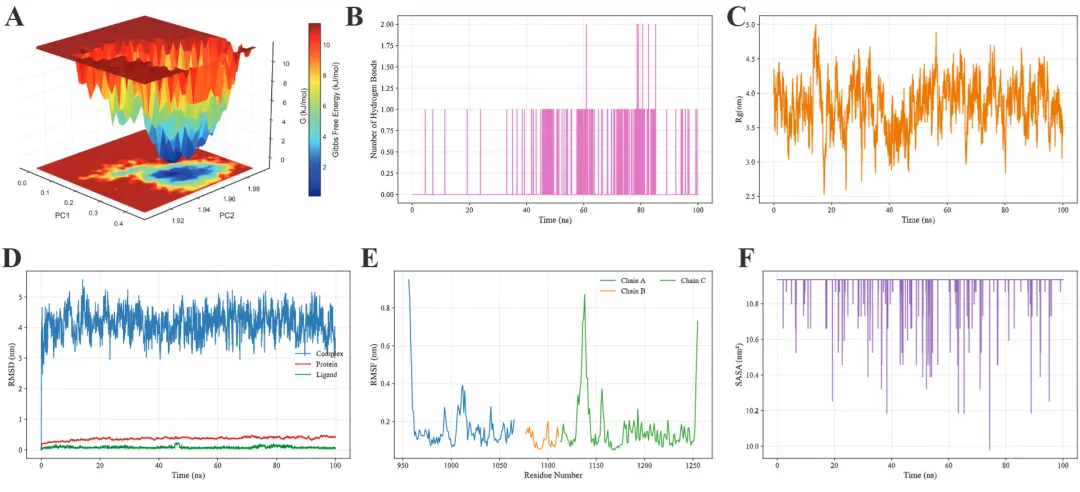
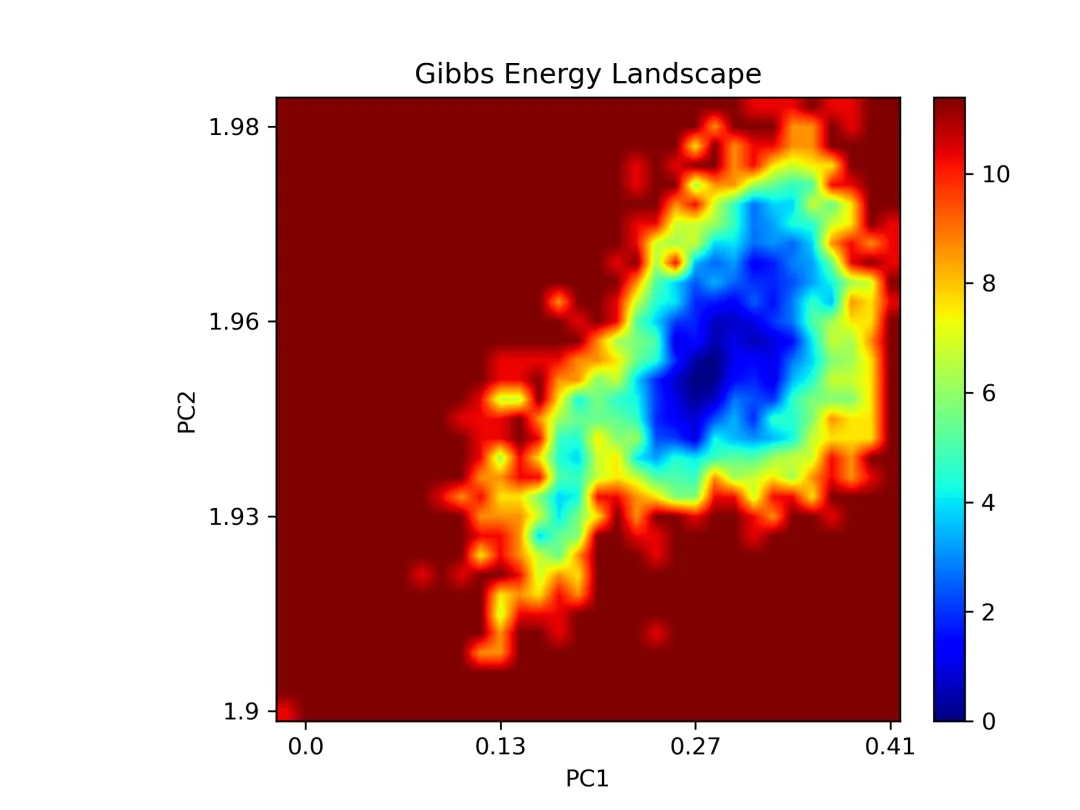
free_energy_2d.png/pdf: 2D自由能景观
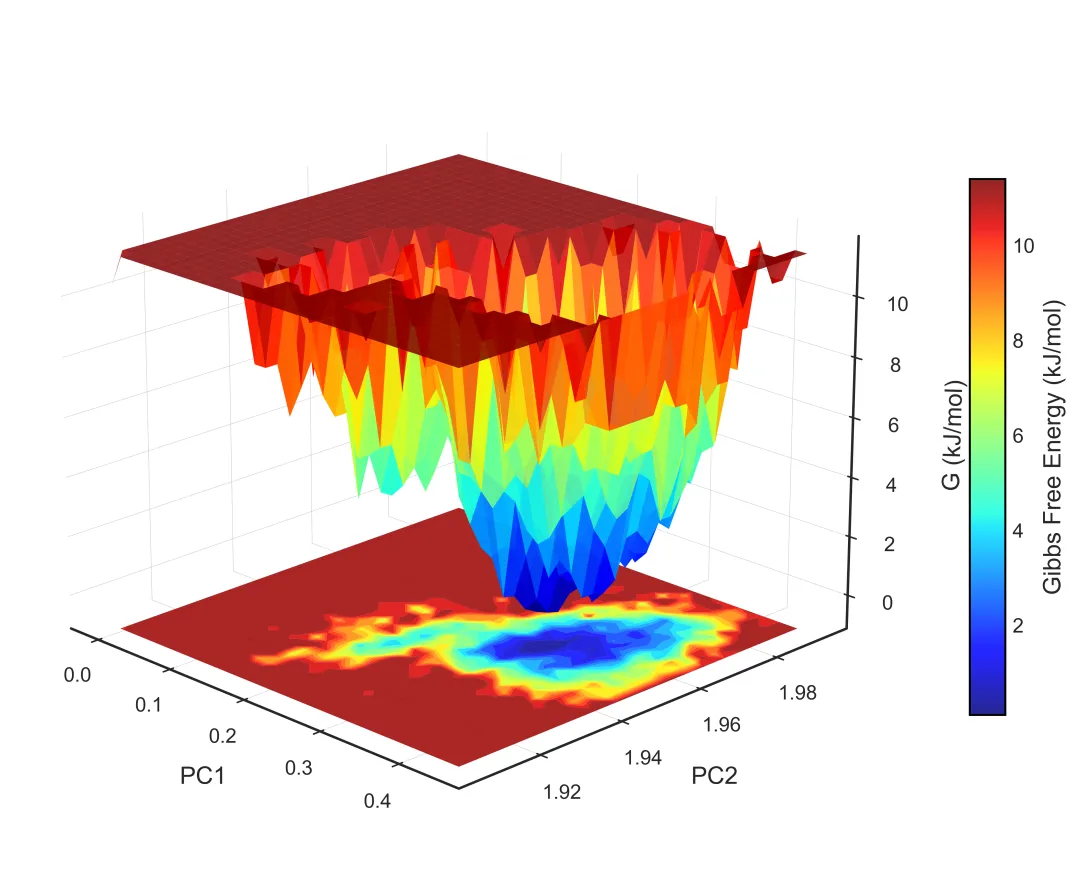
free_energy_3d.png/pdf: 3D自由能景观
contact_map.png/pdf: 接触图
secondary_structure.png/pdf: 二级结构演化
参数调整
修改模拟时长
# md_simulation.py 第127行
steps=5000000# 当前10ns,改为50000000即为100ns
修改温度
# md_simulation.py 第128行
temperature=300# 300K,可改为其他温度
修改保存频率
# md_simulation.py 第129行
report_interval=5000# 每5000步保存一次
GPU使用
确保CUDA可用:
python -c"from openmm import Platform; print(Platform.getPlatformByName('CUDA'))"
GROMACS自动检测GPU,可通过以下命令确认:
gmx mdrun -nb gpu -bonded gpu -pme gpu
常见问题
缺少配体参数: 确保安装了AmberTools和ACPYPE
GPU不可用: 检查CUDA驱动和OpenMM/GROMACS的GPU支持
内存不足: 减小水盒子尺寸或使用更少的溶剂分子
代码:
方法一:OpenMM
md_simulation.py
#!/usr/bin/env python3"""分子动力学模拟主脚本使用OpenMM进行GPU加速的蛋白-配体复合物MD模拟"""import osimport sysfrom openmm import *from openmm.app import *from openmm.unit import *from pdbfixer import PDBFixerimport mdtraj as mdimport numpy as npclass MDSimulation: def __init__(self, protein_pdb, ligand_mol2, output_dir="md_output"): self.protein_pdb = protein_pdb self.ligand_mol2 = ligand_mol2 self.output_dir = output_dir os.makedirs(output_dir, exist_ok=True) def prepare_protein(self): """准备蛋白质:添加氢原子、补全残基、优化结构""" print("准备蛋白质结构...") fixer = PDBFixer(filename=self.protein_pdb) # 查找缺失残基 fixer.findMissingResidues() # 查找缺失原子 fixer.findMissingAtoms() fixer.addMissingAtoms() # 添加氢原子 fixer.addMissingHydrogens(7.0) # 保存处理后的蛋白 self.prepared_protein = f"{self.output_dir}/protein_prepared.pdb" with open(self.prepared_protein, 'w') as f: PDBFile.writeFile(fixer.topology, fixer.positions, f) print(f"✓ 蛋白质已准备完成: {self.prepared_protein}") return self.prepared_protein def prepare_ligand(self): """准备配体:使用GAFF力场参数化""" print("准备配体参数...") # 使用antechamber和parmchk2处理配体 ligand_name = os.path.splitext(os.path.basename(self.ligand_mol2))[0] # 转换为GAFF格式 os.system(f"antechamber -i {self.ligand_mol2} -fi mol2 -o {self.output_dir}/lig.mol2 -fo mol2 -c bcc -at gaff2 -rn LIG") os.system(f"parmchk2 -i {self.output_dir}/lig.mol2 -f mol2 -o {self.output_dir}/lig.frcmod") # 生成配体拓扑 tleap_input = f"""source leaprc.gaff2source leaprc.water.tip3pLIG = loadmol2 {self.output_dir}/lig.mol2loadamberparams {self.output_dir}/lig.frcmodsaveoff LIG {self.output_dir}/lig.libsaveamberparm LIG {self.output_dir}/lig.prmtop {self.output_dir}/lig.inpcrdquit""" with open(f"{self.output_dir}/tleap_lig.in", 'w') as f: f.write(tleap_input) os.system(f"tleap -f {self.output_dir}/tleap_lig.in > {self.output_dir}/tleap_lig.log") print(f"✓ 配体已参数化完成") return f"{self.output_dir}/lig.prmtop", f"{self.output_dir}/lig.inpcrd" def create_complex(self): """创建蛋白-配体复合物""" print("创建复合物系统...") # 使用tleap创建复合物 tleap_complex = f"""source leaprc.protein.ff14SBsource leaprc.gaff2source leaprc.water.tip3ploadoff {self.output_dir}/lig.libloadamberparams {self.output_dir}/lig.frcmodprotein = loadpdb {self.prepared_protein}ligand = loadpdb {self.ligand_mol2}complex = combine {{protein ligand}}# 添加离子中和系统addions complex Na+ 0addions complex Cl- 0# 溶剂化(TIP3P水盒子,距离12埃)solvateBox complex TIP3PBOX 12.0saveamberparm complex {self.output_dir}/complex.prmtop {self.output_dir}/complex.inpcrdsavepdb complex {self.output_dir}/complex.pdbquit""" with open(f"{self.output_dir}/tleap_complex.in", 'w') as f: f.write(tleap_complex) os.system(f"tleap -f {self.output_dir}/tleap_complex.in > {self.output_dir}/tleap_complex.log") print(f"✓ 复合物系统已创建") return f"{self.output_dir}/complex.prmtop", f"{self.output_dir}/complex.inpcrd" def run_simulation(self, prmtop, inpcrd, temperature=300, steps=5000000, report_interval=5000): """运行MD模拟(GPU加速)""" print("开始MD模拟...") # 加载系统 prmtop = AmberPrmtopFile(prmtop) inpcrd = AmberInpcrdFile(inpcrd) # 创建系统 system = prmtop.createSystem( nonbondedMethod=PME, nonbondedCutoff=1.0*nanometers, constraints=HBonds ) # 设置温度和压强 integrator = LangevinMiddleIntegrator(temperature*kelvin, 1/picosecond, 0.002*picoseconds) system.addForce(MonteCarloBarostat(1*bar, temperature*kelvin)) # 使用GPU平台 platform = Platform.getPlatformByName('CUDA') properties = {'CudaPrecision': 'mixed'} # 创建模拟 simulation = Simulation(prmtop.topology, system, integrator, platform, properties) simulation.context.setPositions(inpcrd.positions) # 能量最小化 print("能量最小化...") simulation.minimizeEnergy(maxIterations=1000) # 设置输出 simulation.reporters.append(DCDReporter(f'{self.output_dir}/trajectory.dcd', report_interval)) simulation.reporters.append(StateDataReporter( f'{self.output_dir}/log.txt', report_interval, step=True, potentialEnergy=True, temperature=True, progress=True, remainingTime=True, speed=True, totalSteps=steps, separator='\t' )) # NPT平衡 print("NPT平衡 (100 ps)...") simulation.step(50000) # 生产run print(f"生产MD ({steps} steps)...") simulation.step(steps) # 保存最终结构 positions = simulation.context.getState(getPositions=True).getPositions() PDBFile.writeFile(prmtop.topology, positions, open(f'{self.output_dir}/final.pdb', 'w')) print("✓ MD模拟完成") return f'{self.output_dir}/trajectory.dcd'def main(): # 初始化 md_sim = MDSimulation("protein.pdb", "lig.mol2") # 1. 准备蛋白 prepared_protein = md_sim.prepare_protein() # 2. 准备配体 lig_prmtop, lig_inpcrd = md_sim.prepare_ligand() # 3. 创建复合物 complex_prmtop, complex_inpcrd = md_sim.create_complex() # 4. 运行模拟 trajectory = md_sim.run_simulation( complex_prmtop, complex_inpcrd, temperature=300, steps=5000000, # 10 ns report_interval=5000 ) print("\n✓✓✓ 所有步骤完成!") print(f"轨迹文件: {trajectory}")if __name__ == "__main__": main()
md_analysis.py
#!/usr/bin/env python3"""MD轨迹分析和SCI级别可视化生成RMSD, RMSF, Rg, SASA, 2D/3D能量景观等图"""import mdtraj as mdimport numpy as npimport matplotlib.pyplot as pltfrom matplotlib import cmfrom mpl_toolkits.mplot3d import Axes3Dimport seaborn as snsfrom scipy.spatial.distance import pdist, squareformfrom sklearn.decomposition import PCAimport warningswarnings.filterwarnings('ignore')# SCI级别图表设置plt.rcParams['font.family'] = 'Arial'plt.rcParams['font.size'] = 12plt.rcParams['axes.linewidth'] = 1.5plt.rcParams['xtick.major.width'] = 1.5plt.rcParams['ytick.major.width'] = 1.5class MDAnalysis: def __init__(self, topology, trajectory, output_dir="analysis"): self.traj = md.load(trajectory, top=topology) self.output_dir = output_dir import os os.makedirs(output_dir, exist_ok=True) def calculate_rmsd(self, reference=0, selection='protein'): """计算RMSD""" print("计算RMSD...") atom_indices = self.traj.topology.select(selection) rmsd = md.rmsd(self.traj, self.traj, reference, atom_indices=atom_indices) * 10 # nm to Å time = self.traj.time / 1000 # ps to ns # 绘制RMSD fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, rmsd, linewidth=1.5, color='#2E86AB', alpha=0.8) ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('RMSD (Å)', fontsize=14, fontweight='bold') ax.set_title('Backbone RMSD', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rmsd.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rmsd.pdf', bbox_inches='tight') plt.close() print(f"✓ RMSD图已保存") return rmsd def calculate_rmsf(self, selection='protein and name CA'): """计算RMSF""" print("计算RMSF...") atom_indices = self.traj.topology.select(selection) rmsf = md.rmsf(self.traj, self.traj, atom_indices) * 10 # nm to Å residue_ids = [self.traj.topology.atom(i).residue.resSeq for i in atom_indices] # 绘制RMSF fig, ax = plt.subplots(figsize=(10, 5), dpi=300) ax.plot(residue_ids, rmsf, linewidth=2, color='#A23B72') ax.fill_between(residue_ids, rmsf, alpha=0.3, color='#A23B72') ax.set_xlabel('Residue Number', fontsize=14, fontweight='bold') ax.set_ylabel('RMSF (Å)', fontsize=14, fontweight='bold') ax.set_title('C-alpha RMSF', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rmsf.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rmsf.pdf', bbox_inches='tight') plt.close() print(f"✓ RMSF图已保存") return rmsf def calculate_rg(self, selection='protein'): """计算回旋半径""" print("计算回旋半径...") atom_indices = self.traj.topology.select(selection) rg = md.compute_rg(self.traj, atom_indices=atom_indices) * 10 # nm to Å time = self.traj.time / 1000 # 绘制Rg fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, rg, linewidth=1.5, color='#F18F01', alpha=0.8) ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('Radius of Gyration (Å)', fontsize=14, fontweight='bold') ax.set_title('Radius of Gyration', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) # 添加平均线 ax.axhline(y=np.mean(rg), color='red', linestyle='--', linewidth=1.5, label=f'Mean: {np.mean(rg):.2f} Å', alpha=0.7) ax.legend(frameon=False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rg.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rg.pdf', bbox_inches='tight') plt.close() print(f"✓ Rg图已保存") return rg def calculate_sasa(self, selection='protein'): """计算溶剂可及表面积""" print("计算SASA...") atom_indices = self.traj.topology.select(selection) sasa = md.shrake_rupley(self.traj, mode='residue') total_sasa = np.sum(sasa, axis=1) * 100 # nm^2 to Å^2 time = self.traj.time / 1000 # 绘制SASA fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, total_sasa, linewidth=1.5, color='#06A77D', alpha=0.8) ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('SASA (Ų)', fontsize=14, fontweight='bold') ax.set_title('Solvent Accessible Surface Area', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/sasa.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/sasa.pdf', bbox_inches='tight') plt.close() print(f"✓ SASA图已保存") return total_sasa def free_energy_landscape_2d(self, selection='protein and name CA'): """2D自由能景观图(PCA前两个主成分)""" print("生成2D自由能景观...") atom_indices = self.traj.topology.select(selection) # 提取坐标并展平 xyz = self.traj.atom_slice(atom_indices).xyz n_frames, n_atoms, _ = xyz.shape xyz_flat = xyz.reshape(n_frames, n_atoms * 3) # PCA降维 pca = PCA(n_components=2) pc = pca.fit_transform(xyz_flat) # 计算2D直方图(概率分布) H, xedges, yedges = np.histogram2d(pc[:, 0], pc[:, 1], bins=50) # 转换为自由能 (kT单位) kT = 0.593 # kcal/mol at 300K H[H == 0] = 1e-10 # 避免log(0) free_energy = -kT * np.log(H.T) free_energy -= free_energy.min() # 最小值归零 # 绘制2D等高线图 fig, ax = plt.subplots(figsize=(8, 7), dpi=300) X, Y = np.meshgrid(xedges[:-1], yedges[:-1]) contourf = ax.contourf(X, Y, free_energy, levels=20, cmap='jet') contour = ax.contour(X, Y, free_energy, levels=10, colors='black', linewidths=0.5, alpha=0.4) cbar = plt.colorbar(contourf, ax=ax) cbar.set_label('Free Energy (kcal/mol)', fontsize=12, fontweight='bold') ax.set_xlabel(f'PC1 ({pca.explained_variance_ratio_[0]*100:.1f}%)', fontsize=14, fontweight='bold') ax.set_ylabel(f'PC2 ({pca.explained_variance_ratio_[1]*100:.1f}%)', fontsize=14, fontweight='bold') ax.set_title('2D Free Energy Landscape', fontsize=16, fontweight='bold') plt.tight_layout() plt.savefig(f'{self.output_dir}/free_energy_2d.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/free_energy_2d.pdf', bbox_inches='tight') plt.close() print(f"✓ 2D自由能景观已保存") return pc, free_energy def free_energy_landscape_3d(self, selection='protein and name CA'): """3D自由能景观图""" print("生成3D自由能景观...") atom_indices = self.traj.topology.select(selection) # 提取坐标 xyz = self.traj.atom_slice(atom_indices).xyz n_frames, n_atoms, _ = xyz.shape xyz_flat = xyz.reshape(n_frames, n_atoms * 3) # PCA降维 pca = PCA(n_components=2) pc = pca.fit_transform(xyz_flat) # 计算2D直方图 H, xedges, yedges = np.histogram2d(pc[:, 0], pc[:, 1], bins=50) # 自由能 kT = 0.593 H[H == 0] = 1e-10 free_energy = -kT * np.log(H.T) free_energy -= free_energy.min() # 3D绘图 fig = plt.figure(figsize=(10, 8), dpi=300) ax = fig.add_subplot(111, projection='3d') X, Y = np.meshgrid(xedges[:-1], yedges[:-1]) surf = ax.plot_surface(X, Y, free_energy, cmap='viridis', edgecolor='none', alpha=0.9, linewidth=0, antialiased=True) # 等高线投影 ax.contour(X, Y, free_energy, levels=10, offset=free_energy.min()-2, cmap='coolwarm', linewidths=1.5, alpha=0.6) ax.set_xlabel(f'PC1 ({pca.explained_variance_ratio_[0]*100:.1f}%)', fontsize=12, fontweight='bold', labelpad=10) ax.set_ylabel(f'PC2 ({pca.explained_variance_ratio_[1]*100:.1f}%)', fontsize=12, fontweight='bold', labelpad=10) ax.set_zlabel('Free Energy (kcal/mol)', fontsize=12, fontweight='bold', labelpad=10) ax.set_title('3D Free Energy Landscape', fontsize=14, fontweight='bold', pad=20) # 调整视角 ax.view_init(elev=30, azim=45) cbar = fig.colorbar(surf, ax=ax, shrink=0.5, aspect=5) cbar.set_label('Free Energy (kcal/mol)', fontsize=10, fontweight='bold') plt.tight_layout() plt.savefig(f'{self.output_dir}/free_energy_3d.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/free_energy_3d.pdf', bbox_inches='tight') plt.close() print(f"✓ 3D自由能景观已保存") return pc, free_energy def contact_map(self, selection='protein and name CA', cutoff=0.8): """接触图""" print("生成接触图...") atom_indices = self.traj.topology.select(selection) # 计算平均结构的距离矩阵 avg_traj = self.traj[len(self.traj)//2] # 使用中间帧 distances = md.compute_contacts(avg_traj, scheme='ca')[0][0] * 10 # nm to Å # 重构距离矩阵 n_atoms = len(atom_indices) dist_matrix = squareform(pdist(avg_traj.atom_slice(atom_indices).xyz[0])) residue_ids = [self.traj.topology.atom(i).residue.resSeq for i in atom_indices] # 绘制接触图 fig, ax = plt.subplots(figsize=(8, 7), dpi=300) im = ax.imshow(dist_matrix, cmap='RdYlBu_r', vmin=0, vmax=30) cbar = plt.colorbar(im, ax=ax) cbar.set_label('Distance (Å)', fontsize=12, fontweight='bold') ax.set_xlabel('Residue Number', fontsize=14, fontweight='bold') ax.set_ylabel('Residue Number', fontsize=14, fontweight='bold') ax.set_title('Contact Map', fontsize=16, fontweight='bold') plt.tight_layout() plt.savefig(f'{self.output_dir}/contact_map.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/contact_map.pdf', bbox_inches='tight') plt.close() print(f"✓ 接触图已保存") return dist_matrix def secondary_structure_timeline(self): """二级结构时间演化图""" print("计算二级结构...") dssp = md.compute_dssp(self.traj, simplified=True) # 转换为数值 ss_map = {'C': 0, 'E': 1, 'H': 2} dssp_numeric = np.array([[ss_map[s] for s in frame] for frame in dssp]) time = self.traj.time / 1000 fig, ax = plt.subplots(figsize=(12, 6), dpi=300) im = ax.imshow(dssp_numeric.T, aspect='auto', cmap='Set3', extent=[time[0], time[-1], 0, dssp_numeric.shape[1]]) ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('Residue Number', fontsize=14, fontweight='bold') ax.set_title('Secondary Structure Evolution', fontsize=16, fontweight='bold') # 图例 from matplotlib.patches import Patch legend_elements = [ Patch(facecolor='C', label='Coil'), Patch(facecolor='E', label='β-Sheet'), Patch(facecolor='H', label='α-Helix') ] ax.legend(handles=legend_elements, loc='upper right', frameon=False) plt.tight_layout() plt.savefig(f'{self.output_dir}/secondary_structure.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/secondary_structure.pdf', bbox_inches='tight') plt.close() print(f"✓ 二级结构图已保存") return dsspdef main(): # 加载轨迹 analyzer = MDAnalysis( topology='md_output/complex.pdb', trajectory='md_output/trajectory.dcd' ) # 运行所有分析 print("\n=== 开始分析 ===\n") rmsd = analyzer.calculate_rmsd() rmsf = analyzer.calculate_rmsf() rg = analyzer.calculate_rg() sasa = analyzer.calculate_sasa() pc, fe_2d = analyzer.free_energy_landscape_2d() pc, fe_3d = analyzer.free_energy_landscape_3d() contact = analyzer.contact_map() dssp = analyzer.secondary_structure_timeline() print("\n✓✓✓ 所有分析完成!") print(f"所有图表已保存至 'analysis/' 目录")if __name__ == "__main__": main()
md_simulation_gmx.py
#!/usr/bin/env python3"""使用GROMACS进行MD模拟的Python包装脚本"""import osimport subprocessimport sysclass GMXSimulation: def __init__(self, protein_pdb, ligand_mol2, output_dir="gmx_output"): self.protein_pdb = protein_pdb self.ligand_mol2 = ligand_mol2 self.output_dir = output_dir os.makedirs(output_dir, exist_ok=True) os.chdir(output_dir) def run_cmd(self, cmd): """运行shell命令""" print(f"运行: {cmd}") result = subprocess.run(cmd, shell=True, capture_output=True, text=True) if result.returncode != 0: print(f"错误: {result.stderr}") return result def prepare_protein(self): """准备蛋白质结构""" print("准备蛋白质...") # 使用pdb2gmx添加氢原子并生成拓扑 cmd = f"""gmx pdb2gmx -f ../{self.protein_pdb} -o protein_processed.gro \\ -water tip3p -ff amber99sb-ildn -ignh""" self.run_cmd(cmd) print("✓ 蛋白质已准备") return "protein_processed.gro" def prepare_ligand_acpype(self): """使用ACPYPE准备配体""" print("准备配体(ACPYPE)...") # ACPYPE生成GROMACS拓扑 cmd = f"acpype -i ../{self.ligand_mol2} -n 0 -a gaff2" self.run_cmd(cmd) # 重命名文件 os.system("mv *.acpype/lig_GMX.gro lig.gro") os.system("mv *.acpype/lig_GMX.top lig.top") os.system("mv *.acpype/lig_GMX.itp lig.itp") print("✓ 配体已参数化") return "lig.gro", "lig.top" def create_complex_system(self): """创建复合物系统""" print("创建复合物...") # 合并蛋白和配体 self.run_cmd("cat protein_processed.gro lig.gro > complex_temp.gro") # 编辑拓扑文件 with open("topol.top", 'r') as f: lines = f.readlines() # 在topol.top中添加配体 with open("topol.top", 'w') as f: for line in lines: f.write(line) if "forcefield.itp" in line: f.write('#include "lig.itp"\n') f.write('\nLIG 1\n') # 定义盒子 self.run_cmd("gmx editconf -f complex_temp.gro -o complex_box.gro -c -d 1.2 -bt cubic") # 溶剂化 self.run_cmd("gmx solvate -cp complex_box.gro -cs spc216.gro -o complex_solv.gro -p topol.top") # 添加离子 self.run_cmd("gmx grompp -f ../mdp/ions.mdp -c complex_solv.gro -p topol.top -o ions.tpr -maxwarn 2") self.run_cmd("echo 'SOL' | gmx genion -s ions.tpr -o complex_ions.gro -p topol.top -pname NA -nname CL -neutral") print("✓ 复合物系统已创建") return "complex_ions.gro" def energy_minimization(self): """能量最小化""" print("能量最小化...") self.run_cmd("gmx grompp -f ../mdp/em.mdp -c complex_ions.gro -p topol.top -o em.tpr -maxwarn 2") self.run_cmd("gmx mdrun -v -deffnm em -nb gpu -bonded gpu") print("✓ 能量最小化完成") return "em.gro" def nvt_equilibration(self): """NVT平衡""" print("NVT平衡...") self.run_cmd("gmx grompp -f ../mdp/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr -maxwarn 2") self.run_cmd("gmx mdrun -v -deffnm nvt -nb gpu -bonded gpu -pme gpu") print("✓ NVT平衡完成") return "nvt.gro" def npt_equilibration(self): """NPT平衡""" print("NPT平衡...") self.run_cmd("gmx grompp -f ../mdp/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr -maxwarn 2") self.run_cmd("gmx mdrun -v -deffnm npt -nb gpu -bonded gpu -pme gpu") print("✓ NPT平衡完成") return "npt.gro" def production_md(self): """生产MD""" print("生产MD...") self.run_cmd("gmx grompp -f ../mdp/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md.tpr -maxwarn 2") self.run_cmd("gmx mdrun -v -deffnm md -nb gpu -bonded gpu -pme gpu") print("✓ 生产MD完成") return "md.xtc" def extract_trajectory(self): """提取并转换轨迹""" print("提取轨迹...") # 去除PBC self.run_cmd("echo '1\n0' | gmx trjconv -s md.tpr -f md.xtc -o md_noPBC.xtc -pbc mol -center") # 转换为PDB用于分析 self.run_cmd("echo '1' | gmx trjconv -s md.tpr -f md_noPBC.xtc -o trajectory.pdb -dt 100") print("✓ 轨迹已提取") return "md_noPBC.xtc"def create_mdp_files(): """创建MDP参数文件""" os.makedirs("mdp", exist_ok=True) # ions.mdp with open("mdp/ions.mdp", 'w') as f: f.write("""; ions.mdpintegrator = steepemtol = 1000.0emstep = 0.01nsteps = 5000nstlist = 10""") # em.mdp - 能量最小化 with open("mdp/em.mdp", 'w') as f: f.write("""; em.mdp - Energy minimizationintegrator = steepemtol = 1000.0emstep = 0.01nsteps = 50000nstlist = 10cutoff-scheme = Verletns_type = gridcoulombtype = PMErcoulomb = 1.0rvdw = 1.0pbc = xyz""") # nvt.mdp - NVT平衡 with open("mdp/nvt.mdp", 'w') as f: f.write("""; nvt.mdp - NVT equilibrationintegrator = mdnsteps = 50000dt = 0.002nstxout = 5000nstvout = 5000nstenergy = 5000nstlog = 5000continuation = noconstraint_algorithm = lincsconstraints = h-bondscutoff-scheme = Verletns_type = gridnstlist = 10rcoulomb = 1.0rvdw = 1.0coulombtype = PMEpme_order = 4fourierspacing = 0.16tcoupl = V-rescaletc-grps = Protein_LIG Water_and_ionstau_t = 0.1 0.1ref_t = 300 300pcoupl = nopbc = xyzgen_vel = yesgen_temp = 300gen_seed = -1""") # npt.mdp - NPT平衡 with open("mdp/npt.mdp", 'w') as f: f.write("""; npt.mdp - NPT equilibrationintegrator = mdnsteps = 50000dt = 0.002nstxout = 5000nstvout = 5000nstenergy = 5000nstlog = 5000continuation = yesconstraint_algorithm = lincsconstraints = h-bondscutoff-scheme = Verletns_type = gridnstlist = 10rcoulomb = 1.0rvdw = 1.0coulombtype = PMEpme_order = 4fourierspacing = 0.16tcoupl = V-rescaletc-grps = Protein_LIG Water_and_ionstau_t = 0.1 0.1ref_t = 300 300pcoupl = Parrinello-Rahmanpcoupltype = isotropictau_p = 2.0ref_p = 1.0compressibility = 4.5e-5refcoord_scaling = compbc = xyzgen_vel = no""") # md.mdp - 生产MD with open("mdp/md.mdp", 'w') as f: f.write("""; md.mdp - Production MDintegrator = mdnsteps = 5000000dt = 0.002nstxout = 0nstvout = 0nstfout = 0nstxout-compressed = 5000compressed-x-grps = Systemnstenergy = 5000nstlog = 5000continuation = yesconstraint_algorithm = lincsconstraints = h-bondscutoff-scheme = Verletns_type = gridnstlist = 10rcoulomb = 1.0rvdw = 1.0coulombtype = PMEpme_order = 4fourierspacing = 0.16tcoupl = V-rescaletc-grps = Protein_LIG Water_and_ionstau_t = 0.1 0.1ref_t = 300 300pcoupl = Parrinello-Rahmanpcoupltype = isotropictau_p = 2.0ref_p = 1.0compressibility = 4.5e-5pbc = xyzgen_vel = no""") print("✓ MDP文件已创建")def main(): # 创建MDP文件 create_mdp_files() # 初始化模拟 sim = GMXSimulation("protein.pdb", "lig.mol2") # 运行完整流程 sim.prepare_protein() sim.prepare_ligand_acpype() sim.create_complex_system() sim.energy_minimization() sim.nvt_equilibration() sim.npt_equilibration() sim.production_md() sim.extract_trajectory() print("\n✓✓✓ GROMACS模拟完成!") print("轨迹文件: gmx_output/md_noPBC.xtc")if __name__ == "__main__": main()
md_analysis_gmx.py
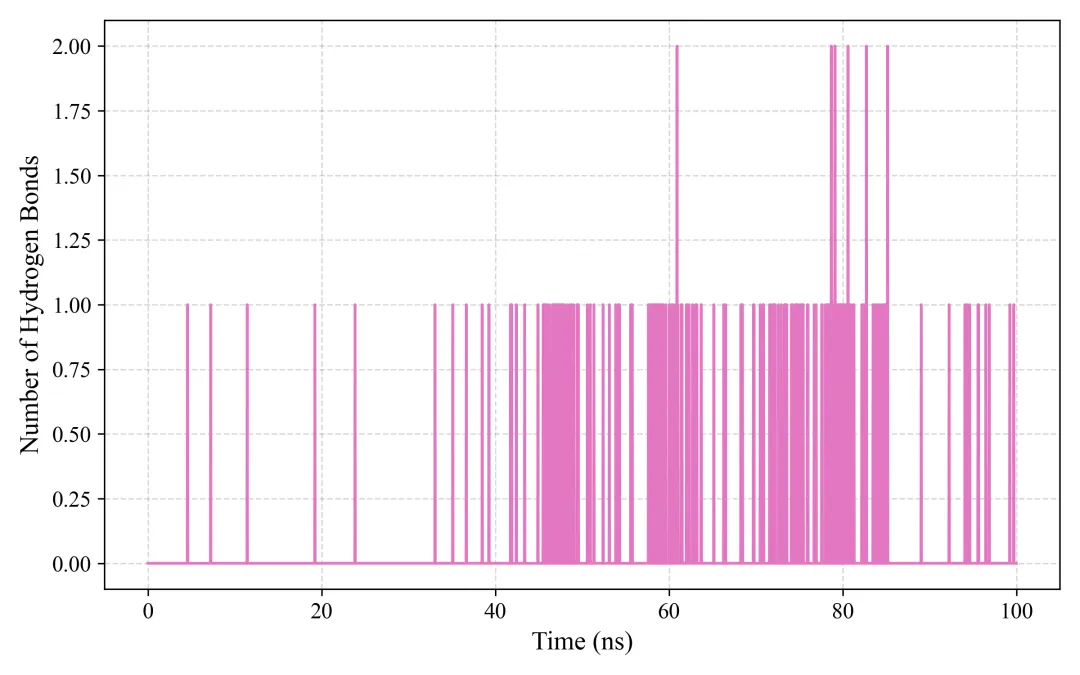
#!/usr/bin/env python3"""GROMACS轨迹分析(使用gmx工具)"""import osimport subprocessimport numpy as npimport matplotlib.pyplot as pltimport seaborn as snsplt.rcParams['font.family'] = 'Arial'plt.rcParams['font.size'] = 12plt.rcParams['axes.linewidth'] = 1.5class GMXAnalysis: def __init__(self, work_dir="gmx_output", output_dir="gmx_analysis"): self.work_dir = work_dir self.output_dir = output_dir os.makedirs(output_dir, exist_ok=True) def run_cmd(self, cmd): """运行命令""" result = subprocess.run(cmd, shell=True, capture_output=True, text=True, cwd=self.work_dir) return result def analyze_rmsd(self): """RMSD分析""" print("计算RMSD...") # 计算backbone RMSD self.run_cmd("echo '4\n4' | gmx rms -s md.tpr -f md_noPBC.xtc -o rmsd.xvg -tu ns") # 读取数据 data = np.loadtxt(f"{self.work_dir}/rmsd.xvg", comments=['@', '#']) time = data[:, 0] rmsd = data[:, 1] * 10 # nm to Å # 绘图 fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, rmsd, linewidth=1.5, color='#2E86AB') ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('RMSD (Å)', fontsize=14, fontweight='bold') ax.set_title('Backbone RMSD', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rmsd.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rmsd.pdf', bbox_inches='tight') plt.close() print("✓ RMSD完成") def analyze_rmsf(self): """RMSF分析""" print("计算RMSF...") # 计算C-alpha RMSF self.run_cmd("echo '3' | gmx rmsf -s md.tpr -f md_noPBC.xtc -o rmsf.xvg -res") # 读取数据 data = np.loadtxt(f"{self.work_dir}/rmsf.xvg", comments=['@', '#']) residues = data[:, 0] rmsf = data[:, 1] * 10 # nm to Å # 绘图 fig, ax = plt.subplots(figsize=(10, 5), dpi=300) ax.plot(residues, rmsf, linewidth=2, color='#A23B72') ax.fill_between(residues, rmsf, alpha=0.3, color='#A23B72') ax.set_xlabel('Residue Number', fontsize=14, fontweight='bold') ax.set_ylabel('RMSF (Å)', fontsize=14, fontweight='bold') ax.set_title('C-alpha RMSF', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rmsf.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rmsf.pdf', bbox_inches='tight') plt.close() print("✓ RMSF完成") def analyze_rg(self): """回旋半径""" print("计算Rg...") # 计算Rg self.run_cmd("echo '1' | gmx gyrate -s md.tpr -f md_noPBC.xtc -o gyrate.xvg") # 读取数据 data = np.loadtxt(f"{self.work_dir}/gyrate.xvg", comments=['@', '#']) time = data[:, 0] rg = data[:, 1] * 10 # nm to Å # 绘图 fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, rg, linewidth=1.5, color='#F18F01') ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('Radius of Gyration (Å)', fontsize=14, fontweight='bold') ax.set_title('Radius of Gyration', fontsize=16, fontweight='bold') ax.axhline(y=np.mean(rg), color='red', linestyle='--', linewidth=1.5, label=f'Mean: {np.mean(rg):.2f} Å', alpha=0.7) ax.legend(frameon=False) ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/rg.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/rg.pdf', bbox_inches='tight') plt.close() print("✓ Rg完成") def analyze_sasa(self): """溶剂可及表面积""" print("计算SASA...") # 计算SASA self.run_cmd("echo '1' | gmx sasa -s md.tpr -f md_noPBC.xtc -o sasa.xvg -tu ns") # 读取数据 data = np.loadtxt(f"{self.work_dir}/sasa.xvg", comments=['@', '#']) time = data[:, 0] sasa = data[:, 1] * 100 # nm^2 to Å^2 # 绘图 fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, sasa, linewidth=1.5, color='#06A77D') ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('SASA (Ų)', fontsize=14, fontweight='bold') ax.set_title('Solvent Accessible Surface Area', fontsize=16, fontweight='bold') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/sasa.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/sasa.pdf', bbox_inches='tight') plt.close() print("✓ SASA完成") def analyze_hbonds(self): """氢键分析""" print("分析氢键...") # 蛋白内部氢键 self.run_cmd("echo '1\n1' | gmx hbond -s md.tpr -f md_noPBC.xtc -num hbond.xvg -tu ns") # 读取数据 data = np.loadtxt(f"{self.work_dir}/hbond.xvg", comments=['@', '#']) time = data[:, 0] hbonds = data[:, 1] # 绘图 fig, ax = plt.subplots(figsize=(8, 5), dpi=300) ax.plot(time, hbonds, linewidth=1.5, color='#D62839') ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('Number of H-bonds', fontsize=14, fontweight='bold') ax.set_title('Hydrogen Bonds', fontsize=16, fontweight='bold') ax.axhline(y=np.mean(hbonds), color='blue', linestyle='--', linewidth=1.5, label=f'Mean: {np.mean(hbonds):.1f}', alpha=0.7) ax.legend(frameon=False) ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/hbonds.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/hbonds.pdf', bbox_inches='tight') plt.close() print("✓ 氢键分析完成") def analyze_dssp(self): """二级结构分析""" print("分析二级结构...") # DSSP分析 self.run_cmd("echo '1' | gmx do_dssp -s md.tpr -f md_noPBC.xtc -o ss.xpm -sc scount.xvg -tu ns") # 读取二级结构统计 data = np.loadtxt(f"{self.work_dir}/scount.xvg", comments=['@', '#']) time = data[:, 0] # 绘图 fig, ax = plt.subplots(figsize=(10, 5), dpi=300) labels = ['Structure', 'Coil', 'β-Sheet', 'β-Bridge', 'Bend', 'Turn', 'α-Helix', '5-Helix', '3-Helix'] colors = ['#E63946', '#F1FAEE', '#A8DADC', '#457B9D', '#1D3557', '#F4A261', '#E76F51', '#2A9D8F', '#264653'] for i in range(1, min(data.shape[1], len(labels))): ax.plot(time, data[:, i], label=labels[i], linewidth=1.5, color=colors[i]) ax.set_xlabel('Time (ns)', fontsize=14, fontweight='bold') ax.set_ylabel('Number of Residues', fontsize=14, fontweight='bold') ax.set_title('Secondary Structure Evolution', fontsize=16, fontweight='bold') ax.legend(frameon=False, loc='upper right') ax.grid(True, alpha=0.3, linestyle='--') ax.spines['top'].set_visible(False) ax.spines['right'].set_visible(False) plt.tight_layout() plt.savefig(f'{self.output_dir}/secondary_structure.png', dpi=300, bbox_inches='tight') plt.savefig(f'{self.output_dir}/secondary_structure.pdf', bbox_inches='tight') plt.close() print("✓ 二级结构分析完成")def main(): analyzer = GMXAnalysis() print("\n=== 开始GROMACS分析 ===\n") analyzer.analyze_rmsd() analyzer.analyze_rmsf() analyzer.analyze_rg() analyzer.analyze_sasa() analyzer.analyze_hbonds() analyzer.analyze_dssp() print("\n✓✓✓ GROMACS分析完成!") print(f"所有图表已保存至 'gmx_analysis/' 目录")if __name__ == "__main__": main()
安装的包:
openmm>=8.0.0mdtraj>=1.9.7numpy>=1.21.0matplotlib>=3.5.0seaborn>=0.12.0scipy>=1.7.0scikit-learn>=1.0.0pdbfixer>=1.8
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
