easySCFr | 一行代码读取空间转录组h5为seurat对象
- 2026-07-02 11:51:36
easySCFr | 一行代码读取空间转录组h5为seurat对象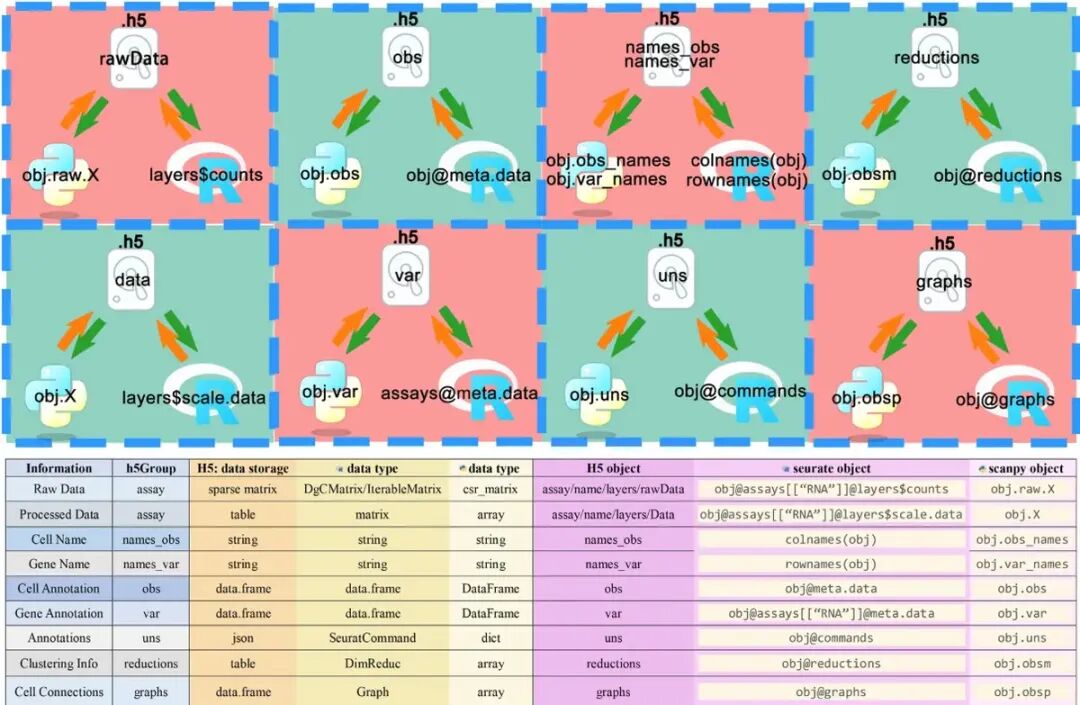
往期回顾 

前面一篇分享了一个读取单细胞数据的工具,有人提到空间转录组是否可用。那么,今天就来分享一个可以快速读取空间转录组h5文件为seurat对象的工具,过程也是相当简单。
library(easySCFr)obj <- loadH5('stxBrain.h5', assay='Spatial', image_name='anterior1', calData=F, calScale=F, calFeatures=F)将h5文件读取为seurat对象只一行代码就可以搞定,如果需要其他软件的格式,可以通过readType参数修改,可选择类型为:Seurat、monocle2、monocle3、cellchat、SingleCellExperiment。cal开头的三个参数默认为开启状态,会进行一些预处理,建议还是关闭吧。
将对象保存为h5文件也是一行代码即可搞定:
saveH5(obj, assay='Spatial', image_name='anterior1', FileName='stxBrain.h5')读取和保存相对重要的两个参数都是assay、image_name,决定了读取或保存哪些数据。并且,读取或者保存时image对象名和键名分别默认为Spatial、Spatial_,如果指定了image_name参数会替换默认值。虽然easySCFr很好用,但在面对多组学数据时就会显得有点力不从心。
anndataR | 一行代码读取h5ad为seurat对象
squidpy | 空间转录组分析软件,scanpy团队出品
--------- The End ---------
基因组
|
转录组
|
表观组
|
单细胞
寻找灵魂的工具人
长按扫码加关注
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

