跟着NC文章源代码学单细胞数据R和python的转换(seurat转adata)
- 2026-06-23 23:01:38
跟着NC文章源代码学单细胞数据R和python的转换(seurat转adata)



文章源代码中将R中的umap信息添加到python 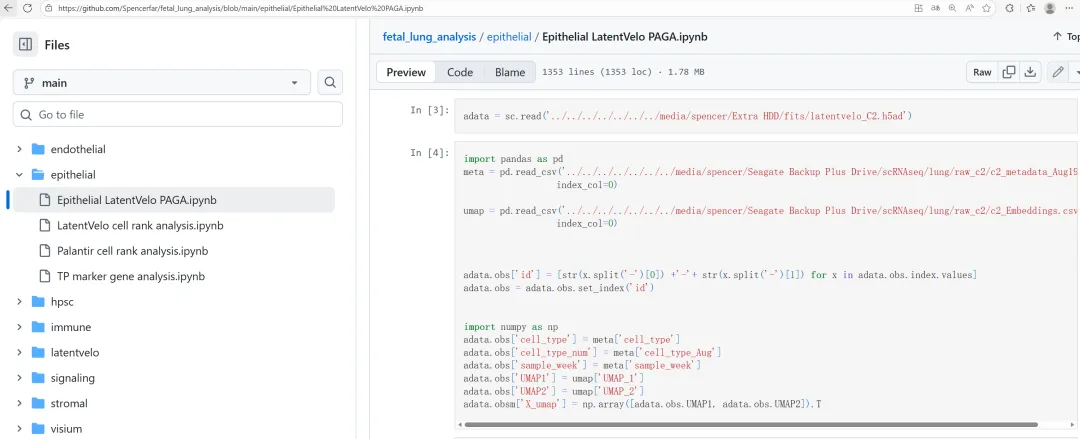

1.加载包并读取数据 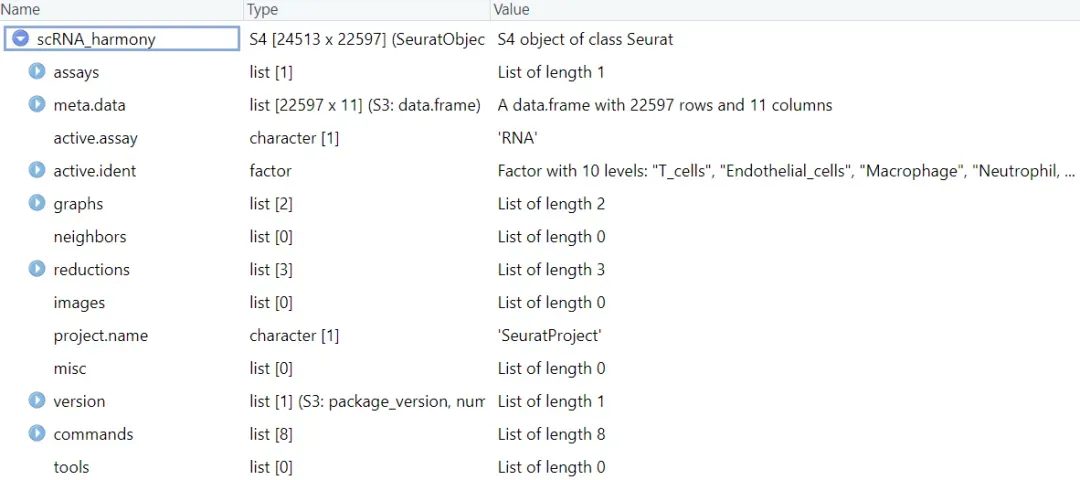
需要画图数据添加作者微信:15623525389 2.基本对象构建 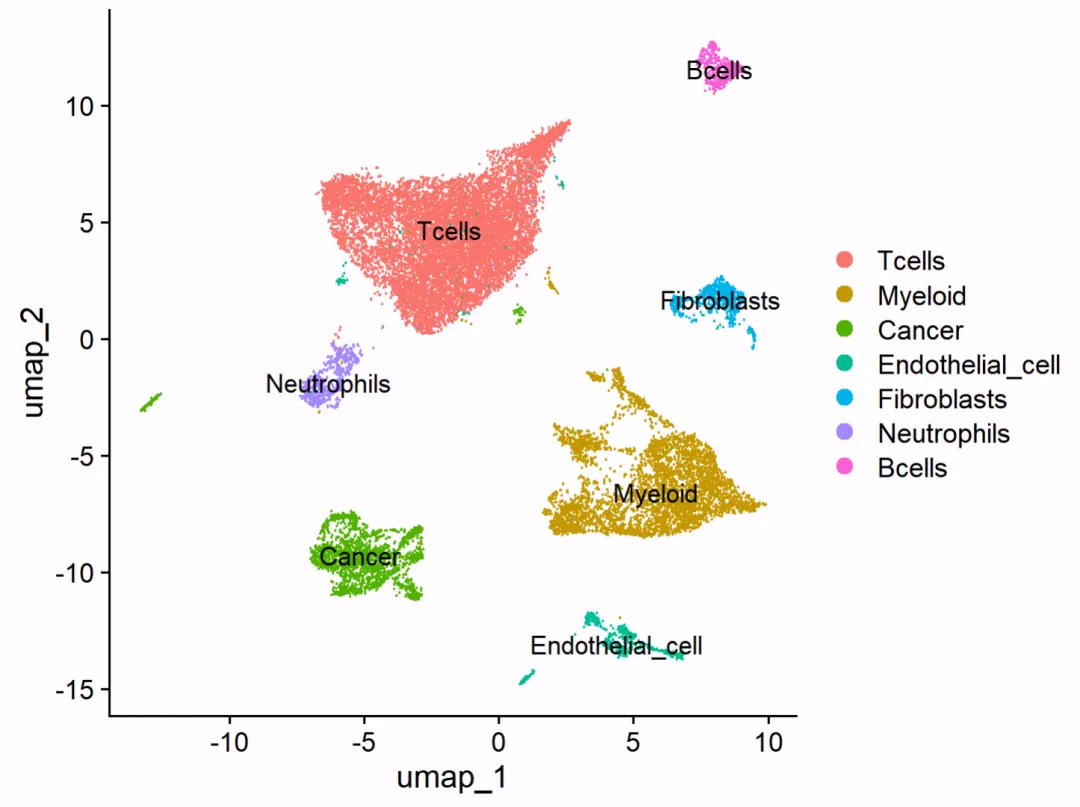
需要画图数据添加作者微信:15623525389 3.自定义函数准备 Seurat → adata 所需文件 4.运行huage_seurat_to_adata函数,提取数据 
数据结果如下 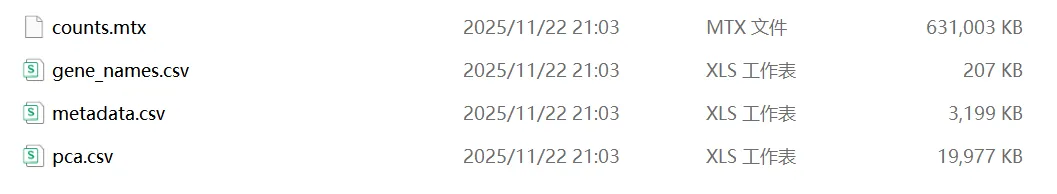
需要画图数据添加作者微信:15623525389

1.在python加载包并读取数据 2.构建函数 huage_seurat_to_adata 3.读取数据 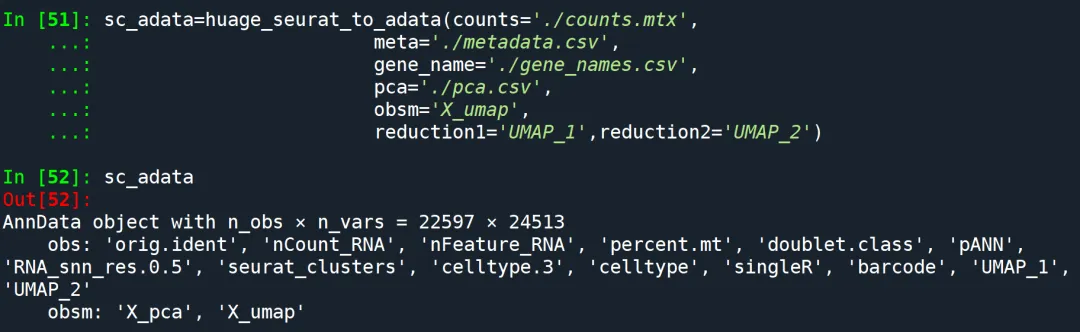
需要画图数据添加作者微信:15623525389 4.绘制 UMAP 图 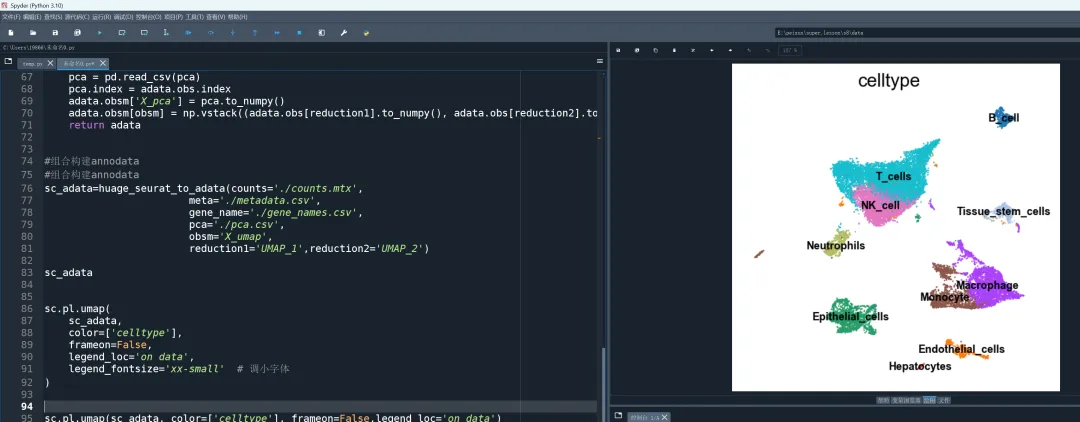
python中的umap图和R中的umap图一模一样 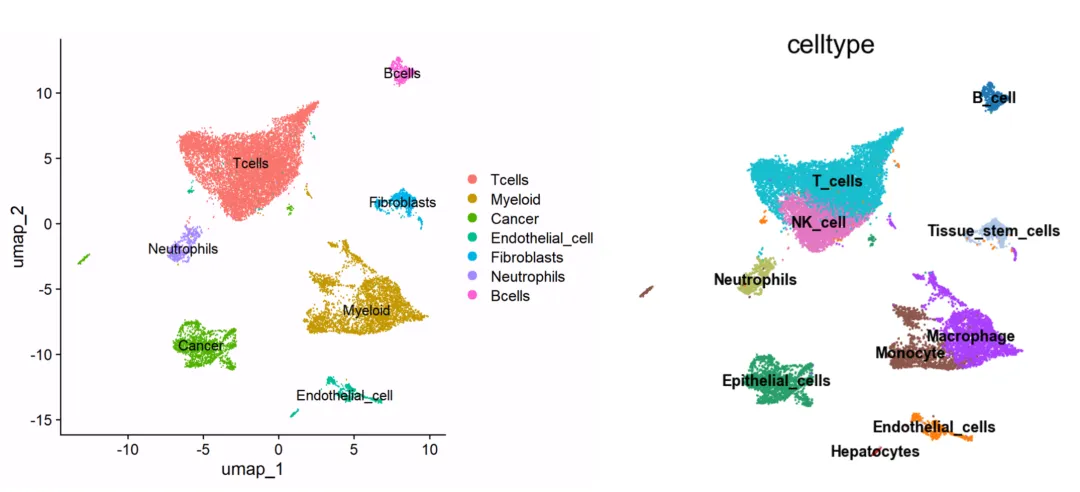
CNS文章单细胞数据分析内容汇总(基础--进阶--python分析) 

最详尽的CNS文章空间转录组数据分析教程 

Nature | 文献
https://www.nature.com/articles/s41467-024-50281-5文章源代码
很多文章的主分析、聚类、分群是在 Seurat(R)里完成的:
已经调了很多参数、过滤了细胞、挑了高变基因
作者在 R 里看图反复调,最终定下来的 UMAP/TSNE 是“人工选过的版本”
这时候如果你在 Python(scanpy、anndata)里再重新跑一遍 UMAP:
用的高变基因、邻居图构建、参数(n_neighbors、min_dist 等)都不一定完全一样
即使你尽量对齐,数值上还是会有微小差异,点的位置/结构会有轻微变化
对于复现论文结果来说,这是不太理想的。所以最干脆的方式就是:把 R 里算好的 UMAP_1 / UMAP_2 直接写进 metadata.csv,然后在 Python 里读出来,放到 adata.obsm['X_umap'] 里用。
R | 处理数据
#系统报错改为英文Sys.setenv(LANGUAGE = "en")##禁止转化为因子options(stringsAsFactors = FALSE)##清空环境rm(list=ls())library(dplyr)library(Matrix)library(reshape2)library(ggplot2)library(ggrepel) #用于注释文本library(magrittr)library(Seurat)library(tidyverse)setwd("E:\\peixun/super.lesson\\s8/")load("anno.scRNA_harmony.rdata")setwd("E:\\peixun/super.lesson\\s8/data/")
## 可视化umap图DimPlot(scRNA_harmony,label = T)
## 预设 cluster 颜色huage_seurat_to_adata <- function(object, # Seurat 对象dimred = c("UMAP", "TSNE"), # 降维方式(单选)assay = "RNA", # 使用的 assay 名slot = "counts", # 使用的表达矩阵 slot(一般是 counts)path = "./" # 文件保存路径(目录)) {# 1. 参数检查与标准化 ---------------------------------------------if(!inherits(object, "Seurat")) {stop("`object` 必须是一个 Seurat 对象。")}dimred <- match.arg(dimred) # 只允许 "UMAP" 或 "TSNE"dimred_lower <- tolower(dimred) # 对应 reductions 名:umap / tsneif(!assay %in% Assays(object)) {stop(paste0("assay '", assay, "' 不在 Seurat 对象中。可用 assay: ",paste(Assays(object), collapse = ", ")))}if(!dir.exists(path)) {dir.create(path, recursive = TRUE)}# 确保 path 末尾有 /if(!grepl("/$", path)) {path <- paste0(path, "/")}# 2. 准备 meta.data ---------------------------------------------seurat_obj <- objectseurat_obj$barcode <- colnames(seurat_obj)meta_df <- seurat_obj@meta.data# 添加降维坐标到 meta.datared_name <- dimred_lower # "umap" 或 "tsne"if(!red_name %in% names(seurat_obj@reductions)) {stop(paste0("Seurat 对象中没有 '", dimred, "' 降维结果 (reductions$", red_name, ")。"))}cell.embeddings <- seurat_obj@reductions[[red_name]]@cell.embeddingsif(dimred == "UMAP") {meta_df$UMAP_1 <- cell.embeddings[, 1]meta_df$UMAP_2 <- cell.embeddings[, 2]} else if (dimred == "TSNE") {meta_df$TSNE_1 <- cell.embeddings[, 1]meta_df$TSNE_2 <- cell.embeddings[, 2]}# 3. 保存 meta.data ---------------------------------------------write.csv(meta_df,file = file.path(path, "metadata.csv"),quote = FALSE,row.names = FALSE)# 4. 保存表达矩阵(counts.mtx) ---------------------------------counts_matrix <- GetAssayData(seurat_obj, assay = assay, slot = slot)# 如果不是稀疏矩阵,转换一下,节省空间if(!inherits(counts_matrix, "dgCMatrix")) {counts_matrix <- as(counts_matrix, "dgCMatrix")}writeMM(counts_matrix,file = file.path(path, "counts.mtx"))# 5. 保存 PCA 信息 ----------------------------------------------if(!"pca" %in% names(seurat_obj@reductions)) {warning("Seurat 对象中没有 PCA 降维结果 (reductions$pca),将跳过 pca.csv。")} else {pca_embed <- seurat_obj@reductions$pca@cell.embeddingswrite.csv(pca_embed,file = file.path(path, "pca.csv"),quote = FALSE,row.names = FALSE)}# 6. 保存 gene 名称 ---------------------------------------------gene_names <- rownames(counts_matrix)write.table(data.frame(gene = gene_names),file = file.path(path, "gene_names.csv"),quote = FALSE,row.names = FALSE,col.names = FALSE)message("✅ seurat_to_adata 完成,文件已保存到: ", normalizePath(path))invisible(path)}
huage_seurat_to_adata(scRNA_harmony,dimred = "UMAP",assay = "RNA",slot = "counts",path = "./")
Python | 处理数据
import pandas as pdimport numpy as npimport scanpy as scimport anndatafrom scipy import iofrom scipy.sparse import coo_matrix, csr_matriximport osos.chdir("E:/peixun\\super.lesson\\s8\\data")sc.settings.verbosity = 0sc.logging.print_versions()sc.settings.set_figure_params(dpi=80, facecolor='white')
def huage_seurat_to_adata(counts, # R 中导出的 counts.mtxmeta, # R 中导出的 metadata.csvgene_name, # R 中导出的 gene_names.csv 或 txtpca, # R 中导出的 pca.csvobsm="X_umap", # 存到 adata.obsm 的键名,比如 "X_umap" 或 "X_tsne"reduction1="UMAP_1", # TSNE_1 或 UMAP_1reduction2="UMAP_2", # TSNE_2 或 UMAP_2):"""将 Seurat 导出的 mtx + metadata + gene_names + pca 转成 AnnData 对象。Parameters----------counts : str or Pathcounts.mtx 的路径(基因 x 细胞)。meta : str or Pathmetadata.csv 的路径,至少包含 'barcode' 列。gene_name : str or Path基因名文件路径(csv/txt,假定第一列是基因名)。pca : str or PathPCA 文件路径(细胞 x PC),行顺序与 metadata 一致。obsm : str嵌入存入 adata.obsm 的键名,比如 "X_umap" 或 "X_tsne"。reduction1 : strmetadata 中低维坐标的第一维列名(如 "UMAP_1" 或 "TSNE_1")。reduction2 : strmetadata 中低维坐标的第二维列名(如 "UMAP_2" 或 "TSNE_2")。Returns-------adata : anndata.AnnData"""# 加载表达矩阵X = io.mmread(counts)# 创建anndataadata = anndata.AnnData(X=X.transpose().tocsr())cell_meta = pd.read_csv(meta)#metadatawith open(gene_name, 'r') as f:gene_names = f.read().splitlines()adata.obs = cell_metaadata.obs.index = adata.obs['barcode']adata.var.index = gene_namespca = pd.read_csv(pca)pca.index = adata.obs.indexadata.obsm['X_pca'] = pca.to_numpy()adata.obsm[obsm] = np.vstack((adata.obs[reduction1].to_numpy(), adata.obs[reduction2].to_numpy())).Treturn adata
## 预设 cluster 颜色sc_adata=huage_seurat_to_adata(counts='./counts.mtx',meta='./metadata.csv',gene_name='./gene_names.csv',pca='./pca.csv',obsm='X_umap',reduction1='UMAP_1',reduction2='UMAP_2')sc_adata
sc.pl.umap(sc_adata, color=['celltype'], frameon=False,legend_loc='on data')强烈推荐,想学生信,复现CNS文章源代码,看👇这个文章:
欢迎关注 | 华哥科研平台
往期精彩内容
亲,写的这么辛苦,记得关注、点赞、打赏哟!
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

