动脉粥样硬化性疾病,如心肌梗死、缺血性中风和外周动脉疾病,仍是全球死亡的主因,尽管降胆固醇药物和药物洗脱支架已取得显著疗效。这一现状凸显了开发新治疗靶点的迫切性。动脉粥样硬化斑块并非均匀分布,而是优先形成于动脉弯曲和分支处。这些区域的血流呈现紊乱特征,内皮细胞承受低幅振荡剪切应力(OSS)。相反,直动脉区域因稳定的层流(伴随高强度单向剪切应力,ULS)而受到保护,后者通过诱导内皮细胞稳态发挥抗动脉粥样硬化作用。血流通过机械传感器和信号转导通路,深刻调控内皮细胞的结构、功能、转录组、表观基因组及代谢,构成疾病发生的地理性基础。
血流紊乱通过内皮重编程驱动疾病进程。研究表明,扰动血流可将动脉内皮细胞从健康表型原位重编程为促动脉粥样硬化表型,这一过程被称为“血流紊乱诱导内皮细胞重编程(FIRE)”。单细胞组学分析揭示,FIRE的特征包括内皮炎症激活、内皮-间质转化(EndMT)、内皮细胞向类免疫细胞转化以及代谢异常。在此病理状态下,内皮屏障功能受损,促进低密度脂蛋白渗透和单核细胞黏附迁移;浸润的免疫细胞分化为巨噬细胞并吞噬氧化脂质形成泡沫细胞,同时血管平滑肌细胞(VSMC)发生合成表型转化、迁移增殖并参与泡沫细胞形成,共同推动脂肪条纹和纤维斑块的发展。尽管高胆固醇血症等全身性风险因素存在,但血流动力学决定了斑块的空间定位。
基于此,美国埃默里大学与佐治亚理工学院生物医学工程系的研究团队在一篇综述中,论述了FIRE的分子机制,即扰动血流如何通过机械传感重编程内皮细胞功能,有望发现调控内皮稳态的关键基因、蛋白及通路,进而开发出阻断血流依赖性促动脉粥样硬化进程的新型疗法。研究成果发表于“Nature reviews. Cardiology”期刊题为“Flow-induced reprogramming of endothelial cells in atherosclerosis”。
动脉粥样硬化斑块优先发生于血流紊乱的血管区域(如颈内动脉分叉侧壁、主动脉弓小弯、左冠前降支近端),其机制与血流动力学特征密切相关。血管内皮暴露于两种关键力学环境,在直动脉区,粘性力主导形成稳定层流,产生高强度单向剪切应力(ULS, ~15 dyn/cm²),维持内皮稳态;而在弯曲/分支处,惯性力引发扰动流,形成低幅振荡剪切应力(OSS, ~±4 dyn/cm²),导致内皮功能障碍。当此类局部血流紊乱与高胆固醇血症、糖尿病等全身风险因素共存时,协同破坏内皮屏障功能、促进炎症及脂质沉积,最终驱动斑块在特定部位的定向发展。血流动力学特征由此成为动脉粥样硬化空间分布的核心决定因素。
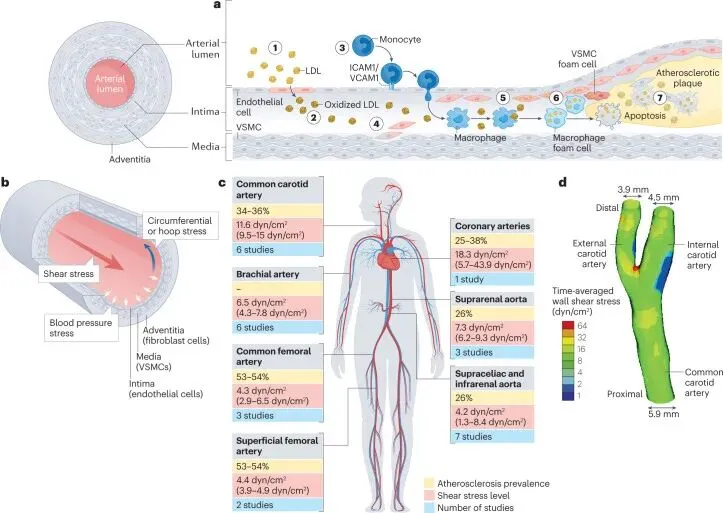
图1 动脉粥样硬化优先发生在血流紊乱的部位。
动物模型证实血流紊乱驱动动脉粥样硬化。部分颈动脉结扎(PCL)模型通过手术结扎小鼠左颈动脉分支,诱导低幅振荡剪切应力(OSS),使高胆固醇饮食的Apoe⁻/⁻小鼠在2-3周内全段动脉快速形成斑块,而对侧稳定血流区域保持健康,提供对照。收缩性颈动脉袖带模型则单动脉内呈现三重力学梯度,近袖带区低稳流致易损斑块,袖带内高稳流无病变,远段扰动流形成稳定斑块,验证剪切应力模式与斑块性质的因果关联。斑马鱼模型通过基因操作联合血流干预,进一步证实血流紊乱协同高胆固醇血症触发早期动脉粥样硬化事件。这些模型为血流致病机制提供了的体内证据。
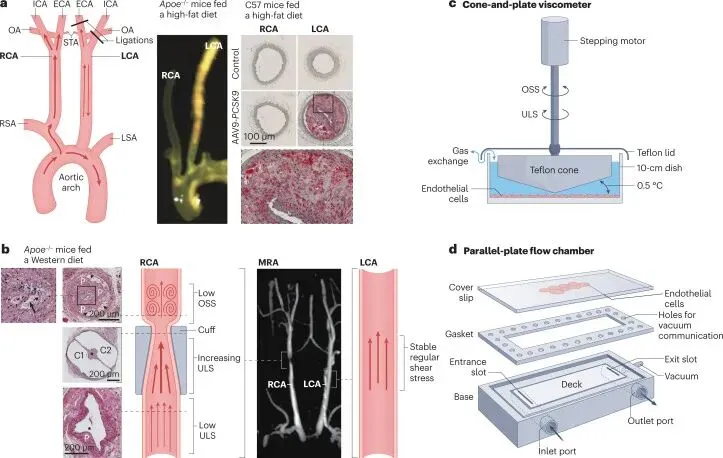
图2 血流紊乱诱发的动脉粥样硬化模型。
血流通过剪切应力调控内皮功能。稳定层流(如直动脉区~15 dyn/cm²剪切应力)维持内皮稳态,增强屏障功能(通过occludin、VE-cadherin调控紧密/粘附连接),诱导保护性自噬(Sirtuin1/FOXO1通路),抑制糖酵解代谢。相反,血流紊乱(如弯曲区~±4 dyn/cm²振荡应力)触发内皮功能障碍:增加通透性、促进糖酵解及线粒体裂变、升高ROS(经BMP4-NADPH氧化酶通路),诱导炎症(激活NF-κB,上调VCAM1/ICAM1)和细胞转分化(EndMT)。形态上,稳定流使内皮细胞沿血流方向梭形排列,而紊乱流导致多边鹅卵石状无序排列,伴随肌动球蛋白骨架重组。这些血流依赖性机制共同决定内皮屏障完整性、氧化应激水平及炎症状态,成为动脉粥样硬化的核心调控环节。
内皮细胞通过机械传感器网络将血流信号转化为生物学响应。顶端表面分布着PIEZO1钙通道、NOTCH1受体等膜蛋白及糖萼等结构,其中PIEZO1介导剪切应力依赖性钙内流调控细胞排列,NOTCH1则维持连接完整性并抑制动脉粥样硬化(其与PIEZO1的互作需进一步验证);细胞连接处的PECAM1/VE-cadherin/VEGFR2复合物通过PI3K-AKT通路传递稳定流信号,促进eNOS活化;基底面的整合素则感知细胞外基质力学变化。值得注意的是,稳定流与扰动流可短暂激活相似早期通路(如NF-κB),但前者仅瞬时激活不引发炎症,后者则导致持续信号传导驱动病理进程。这种差异机制尚不明确,可能与体外研究中静态细胞突受剪切力的适应性反应有关。
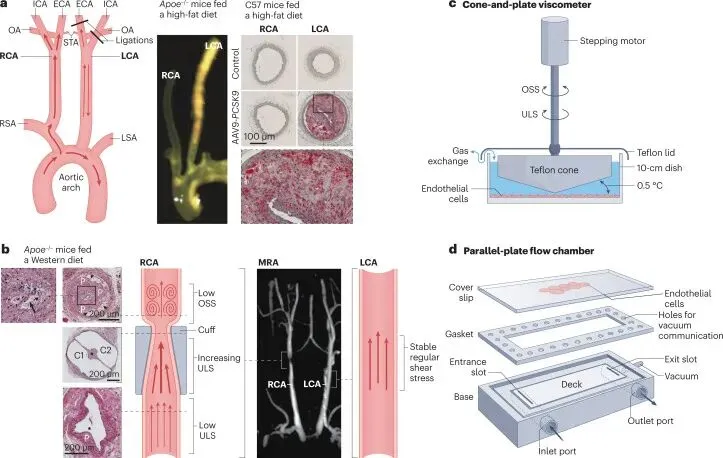
图3 内皮细胞中的机械传感器和机械信号转导途径。
PIEZO1介导血流依赖的双向内皮响应。在稳定层流中,PIEZO1感知剪切应力触发钙内流,诱导ATP释放并激活P2Y2/Gαq/11-AKT-eNOS通路,促进一氧化氮生成,发挥抗动脉粥样硬化及血管舒张作用。相反,在血流紊乱区域,PIEZO1通过Gαq/11激活整合素,进而驱动粘着斑激酶(FAK)依赖的NF-κB信号持续活化,导致内皮炎症和促动脉粥样硬化反应。遗传学证据显示,Ldlr⁻/⁻小鼠内皮特异性敲除Piezo1可显著抑制扰动流区域的斑块发展,证实其促动脉粥样硬化功能。这种同一机械传感器在不同血流模式下介导相反生物学效应的现象,凸显了血流动力学微环境对内皮命运的决定性调控。
Plexin D1作为环境依赖的双向机械传感器。在稳定层流中,Plexin D1介导保护性信号,激活eNOS并诱导转录因子KLF2/KLF4表达,促进内皮细胞排列及抗动脉粥样硬化反应;而在血流紊乱时,它转而驱动促动脉粥样硬化通路,上调VCAM1/CCL2等炎症因子。遗传学研究揭示其功能双向性,内皮特异性敲除Plexin D1基因可抑制扰动流区域的斑块形成,但意外加剧稳定流区域的病变发展。这种"同一分子在不同力学环境下发挥相反作用"的特性,凸显血流模式对机械传感器功能的决定性调控,为靶向治疗提供复杂新视角。
连接机械感觉复合体(PECAM1/VE-cadherin/VEGFR)在血流响应中具双向作用。稳定层流通过该复合体激活PI3K-AKT-eNOS通路,促进内皮保护性一氧化氮生成;同时诱导整合素αvβ3和短暂NF-κB活化,但不引发炎症。在血流紊乱区域,同一复合体(尤其PECAM1)介导持续NF-κB信号传导,驱动内皮炎症和动脉粥样硬化。Pecam1敲除小鼠的扰动流区域内皮炎症及斑块形成显著抑制,证实PECAM1是促动脉粥样硬化的核心机械传感器。这种"同一复合体→相反病理结局"的现象,凸显血流动力学微环境通过分子背景重编程复合体功能的机制。
整合素通过RHO GTP酶介导血流依赖的内皮表型转换。在稳定层流中,整合素激活使RHOA失活,促进YAP在细胞质中的磷酸化(Ser127/Ser381),维持抗动脉粥样硬化内皮表型;同时通过RAC激活组装连接机械感觉复合物,并诱导CDC42极化调控细胞骨架。相反,在血流紊乱时,α5β1/αvβ3整合素与纤连蛋白协同驱动促炎信号,激活PAK激酶,触发NF-κB和YAP核转位,导致炎症反应。这种双向调控机制揭示整合素-RHO轴如同分子开关,响应力学环境变化决定内皮细胞命运,稳定流中维持稳态,扰动流中转向病理进程。
流量敏感转录因子构成内皮表型转换枢纽。稳定层流通过MEKK3-MEK5-ERK5级联激活主调控因子KLF2/KLF4,驱动抗炎、抗栓基因表达,并竞争性抑制NF-κB(经由CBP-p300争夺);而血流紊乱则持续活化NF-κB,上调VCAM1/ICAM1及HIF1α(促进糖酵解),并诱导YAP/TAZ核转位介导炎症与细胞骨架重构。SOX13在扰动流中被显著抑制,导致CCL5/CXCL10等趋化因子暴发,放大内皮炎症。这种转录网络的双向调控,KLF2/4维持稳态 vs NF-κB/YAP/HIF1α驱动病变,将机械信号转化为长期内皮表型编程,奠定动脉粥样硬化的转录基础。
单细胞组学揭示扰流诱导内皮重编程(FIRE)的时空动态。通过scRNA-seq/scATAC-seq分析小鼠部分颈动脉结扎模型,突破传统组学的细胞局限,扰动流区域(LCA)出现新型内皮亚群(E6/E8),取代稳态亚群(E2-E4),伴随VSMC/成纤维细胞/免疫细胞浸润。E8亚群显著表达VSMC标记(Acta2/Tagln)、免疫标记(Cd74/H2基因)及EndMT/EndIT特征基因,经伪时间轨迹分析证实其由健康内皮转分化而来。体外实验进一步验证,内皮细胞在扰动流下独立诱导SNAI1(EndMT)和C1QC(EndIT表达,排除免疫细胞干扰。这种血流紊乱驱动的原位内皮重编程(FIRE),以内皮获得间质/免疫样表型为核心特征,构成动脉粥样硬化的细胞起源。
单细胞组学突破性揭示FIRE机制。传统批量转录/表观组学因免疫细胞污染难以识别扰动流下内皮特异性高表达基因。scRNA-seq/scATAC-seq技术以单细胞分辨率解析小鼠颈动脉模型,发现扰动流区域(LCA)出现新型内皮亚群E6/E8,取代健康亚群(E2-E4),E8细胞高表达VSMC标记(Acta2/Tagln)、免疫标记(Cd74/H2基因)及EndMT/EndIT特征基因,体外内皮模型中,扰动流独立诱导SNAI1(EndMT)和C1QC(EndIT)表达,排除免疫细胞干扰。血流紊乱驱动内皮原位重编程(FIRE),以内皮获得间质/免疫样表型为核心特征,为动脉粥样硬化起源提供细胞学基础。
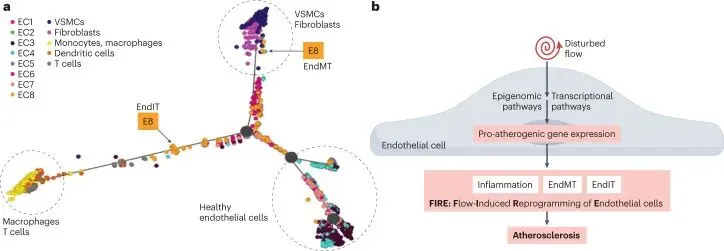
图4 单细胞 RNA 测序揭示扰流诱导的内皮细胞重编程。
靶向血流敏感通路开辟抗动脉粥样硬化新策略。基于CANTOS试验证实炎症靶向治疗的可行性,调控FIRE(内皮炎症/EndMT/EndIT)相关分子成为新兴方向,激活稳定流诱导的动脉粥样硬化保护因子,如KLF2/KLF4(占50%保护基因),他汀类可诱导其表达,桦木酸通过ERK5-MEF2C通路激活KLF2-eNOS;重组KLK10蛋白疗法已证实可抑制斑块;阻断扰动流效应分子,如HIF1α抑制剂PX-478、miR-92a/712拮抗剂、DNMT抑制剂5-aza-2'-脱氧胞苷,均在小鼠模型中显著缓解病变。
总之,血流剪切应力通过机械传感-转录重编程轴双向调控动脉粥样硬化。内皮细胞通过PIEZO1、整合素等机械传感器将力学信号转化为胞内应答,经NF-κB/KLF2等转录因子调控表观基因组与转录组,最终决定内皮表型。稳定流维持抗动脉粥样硬化稳态(屏障完整/抗炎/代谢平衡),而扰动流触发 FIRE(血流诱导内皮重编程),以炎症激活、EndMT(间质转化)及EndIT(免疫样转化)为特征的三位一体病理表型。单细胞组学(scRNA/scATAC-seq)突破性揭示FIRE的时空动态演化,证实内皮原位转分化是斑块形成的细胞起源。靶向FIRE相关分子(如增强KLF2/KLF4保护轴或抑制HIF1α/DNMT损伤轴)有望成为降脂治疗外的补充策略,为破解残余风险提供新方向。
参考文献:Tamargo IA, Baek KI, Kim Y, Park C, Jo H. Flow-induced reprogramming of endothelial cells in atherosclerosis. Nat Rev Cardiol. 2023 Nov;20(11):738-753. doi: 10.1038/s41569-023-00883-1. Epub 2023 May 24. PMID: 37225873; PMCID: PMC10206587.原文链接:https://pubmed.ncbi.nlm.nih.gov/37225873/或点击阅读原文
图片来源:所有图片均来源于参考文献
小编旨在分享、学习、交流生物科学等领域的研究进展。如有侵权或引文不当请联系小编修正。如有任何的想法以及建议,欢迎联系小编。感谢各位的浏览以及关注!






























 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
