python实战直播学习:顶刊杂志的单细胞数据(GSE154826)分析
- 2026-07-03 22:03:09
python基本部分学完了之后,见专辑《python生信笔记》,就需要开始进行大量的数据分析实战练习进行掌握了。今天来学习一篇顶刊杂志的单细胞数据处理,使用python!
note:这个数据主要来自最近开一个直播进行实战训练,详细情况以及进群方式见:python单细胞数据分析实战直播交流群
整个直播计划见文档:https://www.yuque.com/xinnigegui-uqv7c/nneog4/ecg5ec5hscuiudqq
此外,我们生信技能树每月都有一期0基础的生信入门培训班,对新手超级友好,有超贴心的各种答疑服务,3月份的在这里:生信入门&数据挖掘线上直播课2026年3月班。快来添加小助手报名咨询吧。数据背景
今天分析直播的那篇文献里面里面提到的 Maier, Merad et al. 数据,来自文献 10.1016/j.ccell.2021.10.009。文献标题为《Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification》。2021 年12月13号发表在 Cancer Cell 杂志(IF=38.585/Q1)上。
数据集为:data/12_input_adatas/maier_merad_2020_nsclc.h5ad,在文献里面的下载地址为 :https://zenodo.org/records/7227571,在里面的压缩包 input_data.tar.xz 中。
直播文献里面的数据全部在文献的table S1中:
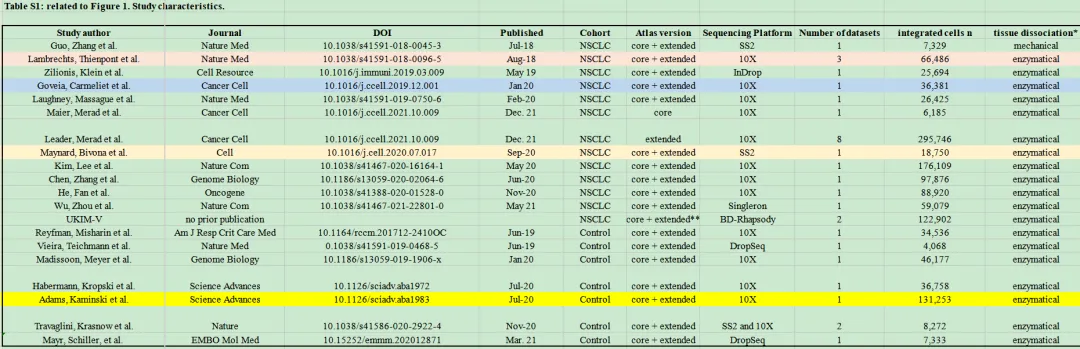
Maier_Merad_2020_NSCLC 简介
本文对 35 例早期 NSCLC 病灶的 361,929 个细胞进行单细胞 RNA 测序,鉴定出一个由 PDCD1+CXCL13+ 活化 T 细胞、IgG+ 浆细胞和 SPP1+ 巨噬细胞构成的细胞模块,命名为肺癌活化模块(LCAMhi)。并在多个 NSCLC 队列中验证了 LCAMhi 的富集特征,并通过配对 CITE-seq 技术建立了识别 LCAMhi 病灶的抗体组合。研究发现 LCAM 的存在与总体免疫细胞含量无关,但与 TMB、癌睾抗原及 TP53 突变显著相关。即使在 TMB 高于中位值的患者中,高基线 LCAM 评分仍与 NSCLC免疫治疗增强应答相关,提示免疫细胞组成虽与 TMB 相关,但可能作为免疫治疗应答的非冗余生物标志物。
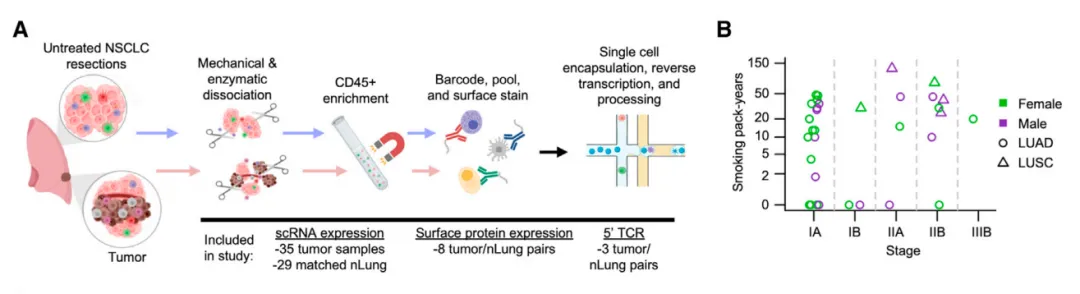
代码:https://github.com/effiken/Leader_et_al
本文数据:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154826
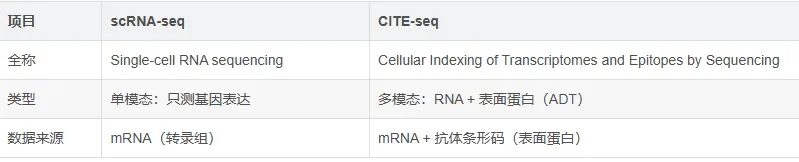
此外,看了下 CITE-seq 与 scRNA-seq的区别。
这个研究的数据用的比较复杂,每个图用了哪几个样本可以参考文献的 Table S7.
这里我就直接用直播的那篇图谱文献里面的h5ad文件了:maier_merad_2020_nsclc.h5ad
数据的百度链接可以在语雀文档里面找到:https://www.yuque.com/xinnigegui-uqv7c/nneog4/ecg5ec5hscuiudqq
0.加载python模块
还是老环境 conda 小环境sc2,缺啥在里面安装就好了:
# %%# %load_ext autoreload# %autoreload 2import scanpy as scimport numpy as npimport pandas as pdimport scipy.sparse as spfrom scanpy_helpers.annotation import AnnotationHelperfrom nxfvars import nxfvarssc.set_figure_params(figsize=(5, 5))1.数据读取
读取上面的h5ad文件,查看里面的各种信息:
../data/12_input_adatas/maier_merad_2020_nsclc.h5ad
# 加载数据adata = sc.read_h5ad(filename="../data/12_input_adatas/maier_merad_2020_nsclc.h5ad" )adataAnnData object with n_obs × n_vars = 6506 × 33660 obs: 'sample', 'annot', 'tissue', 'patient', 'condition', 'origin'
看一下数据其他的信息:
# 看列名adata.obs.columns.to_list()# ['sample', 'annot', 'tissue', 'patient', 'condition', 'origin']adata.obs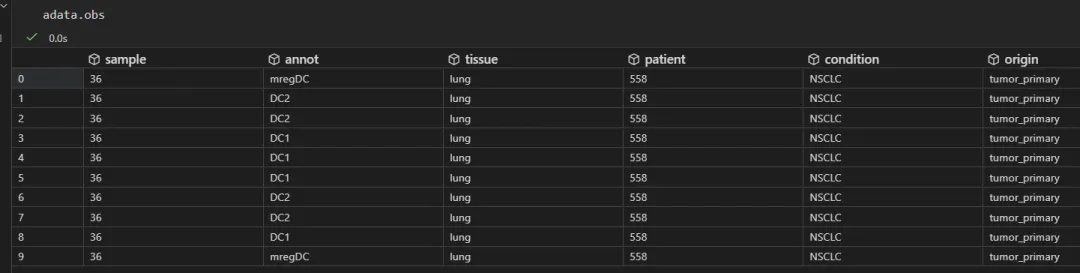
样本数,注释信息:这里看着只用的DC亚群的数据。
adata.obs['sample'].value_counts()adata.obs['annot'].value_counts()adata.obs['tissue'].value_counts()adata.obs['patient'].value_counts()adata.obs['condition'].value_counts()adata.obs['origin'].value_counts()看一些简单的信息:
#表达矩阵里的数值范围np.min(adata.X),np.max(adata.X)#过滤前adata.X.shapeadata[0:6, ['CD3D','TCL1A','MS4A1']].to_df()adata.var.head()# 查看细胞名adata.obs_names[:10]2.数据过滤
首先还是计算每个细胞中线粒体基因,核糖体基因,红血细胞基因表达比例:
# 计算线粒体基因比例adata.var['mt']=adata.var_names.str.startswith('MT-')# 计算核糖体基因比例adata.var['ribo']=adata.var_names.str.match('^RP[SL]')# 计算红血细胞基因比例adata.var['hb']=adata.var_names.str.match('^HB[^(P)]')# 计算sc.pp.calculate_qc_metrics(adata,qc_vars=['mt','ribo','hb'],percent_top=None,log1p=False,inplace=True)# 质控adata.obs.head()# 每个基因在多少个细胞中表达adata.var.head()# 绘制小提琴图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (11, 10)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="sample", jitter=0.4,rotation=90,multi_panel=True,#stripplot=False, # remove the internal dots#inner="box", # adds a boxplot inside violins )过滤前的小提琴信息如下:
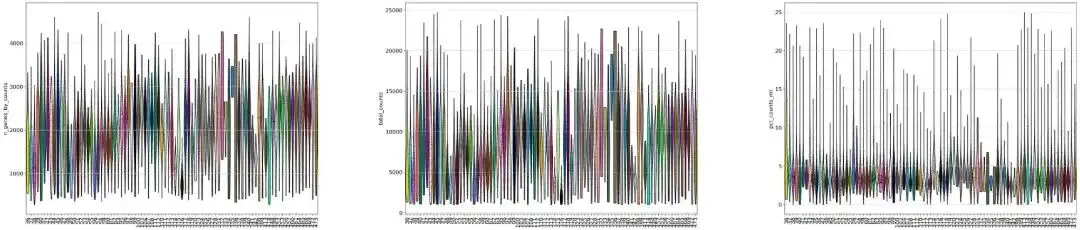
直播文献里面给的过滤参数:
这个过滤指标我每次看到都觉得很随意和主观!!!
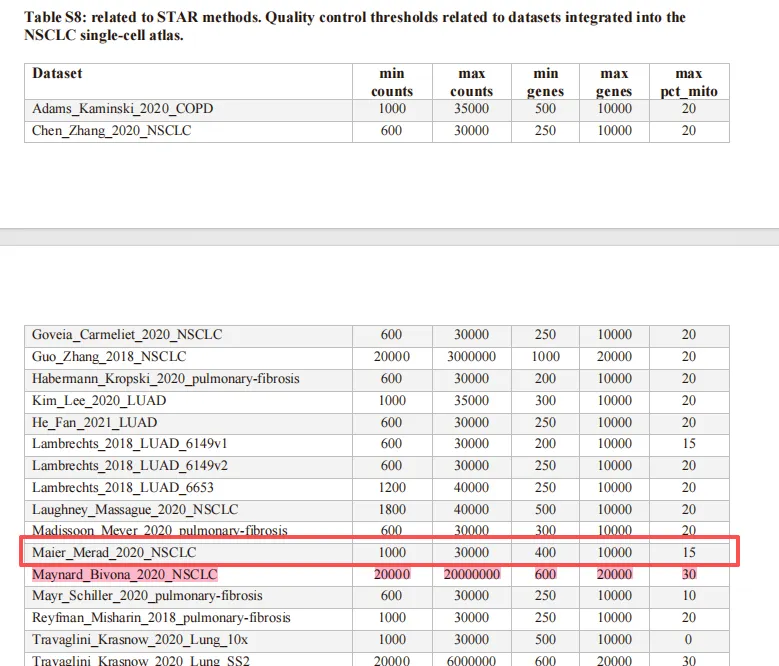
按照文献里面给的看看:
## 过滤前的数据adata.shape# (6506, 33660)## 过滤# 方法2:一次性过滤所有条件(更高效)adata = adata[ (adata.obs['n_genes_by_counts'] > 400) & (adata.obs['n_genes_by_counts'] < 10000) & (adata.obs['total_counts'] > 1000) & (adata.obs['total_counts'] < 30000) & (adata.obs['pct_counts_mt'] < 15), :].copy()## 过滤后的数据adata.shape# (6387, 33660)一共过滤掉了 119个细胞。
过滤后的小提琴图如下:
from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (9, 10)}): sc.pl.violin( adata, ['n_genes_by_counts','total_counts','pct_counts_mt'], groupby="sample", jitter=0.4,rotation=90,multi_panel=True,#stripplot=False, # remove the internal dots#inner="box", # adds a boxplot inside violins )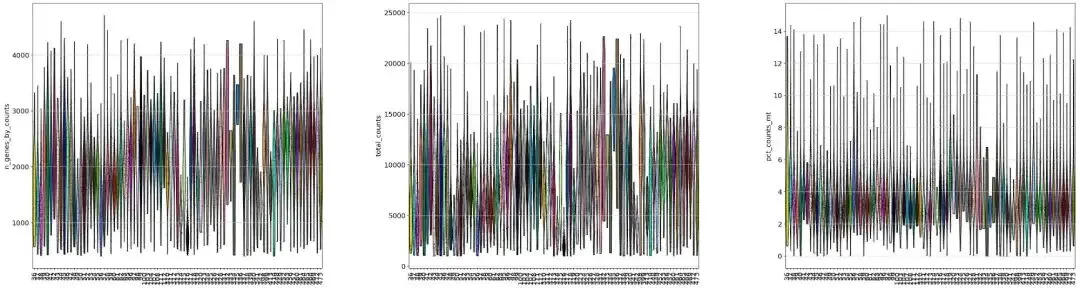
3.降维聚类
基本上都是scanpy的标准流程了:这里细胞数不多跑起来比较快。
# 首先将数据矩阵标准化(校正文库大小):sc.pp.normalize_total(adata,target_sum=1e4)# 对数据进行logsc.pp.log1p(adata)adata.raw = adata.copy()# 高变基因sc.pp.highly_variable_genes(adata)# 归一化sc.pp.scale(adata)# pca分析sc.tl.pca(adata,use_highly_variable=True)# harmony整合sc.external.pp.harmony_integrate(adata,'sample')# 聚类sc.pp.neighbors(adata,use_rep='X_pca_harmony',n_pcs=50)sc.tl.umap(adata)# Using the igraph implementation and a fixed number of iterations can be significantly faster, especially for larger datasetssc.tl.leiden(adata, flavor="igraph", n_iterations=2,resolution=0.5)adata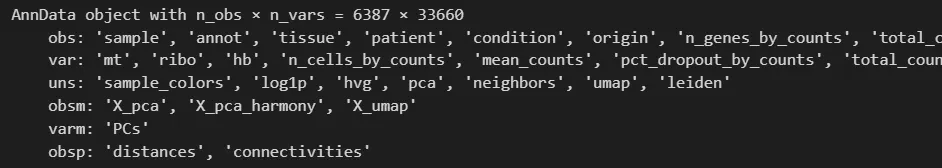
跑完了后可以看到anndata里面多了很多信息,如果你对这个数据结构还不熟悉,看看这个帖子:做单细胞和空转必须了解的AnnData数据结构。
看下umap图
群里经常看到有人问:怎么判断自己的整合效果好不好?问下自己看看。
下面是聚类结果umap:
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (6.5, 6.2)}): sc.pl.umap(adata, color=["leiden"], legend_loc="on data", size=10)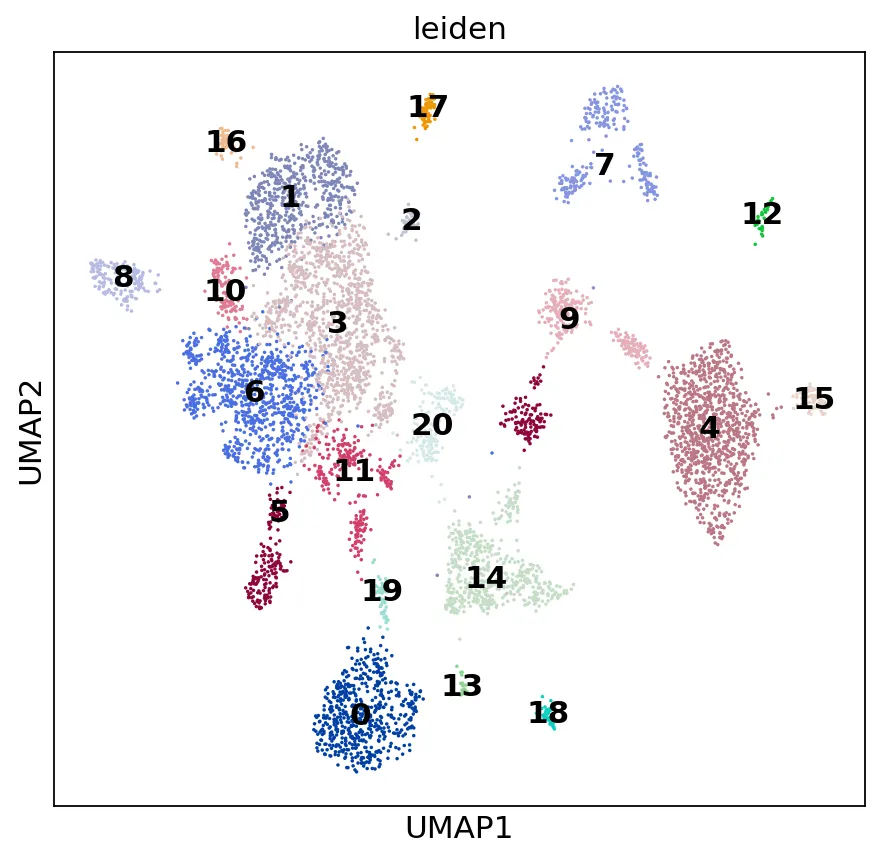
下面是sample上色的聚类结果umap:不同样本均匀分散在各大群里。
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (8, 7)}): sc.pl.umap(adata, color=["sample"], size=10) # 样本太多了所以是灰色?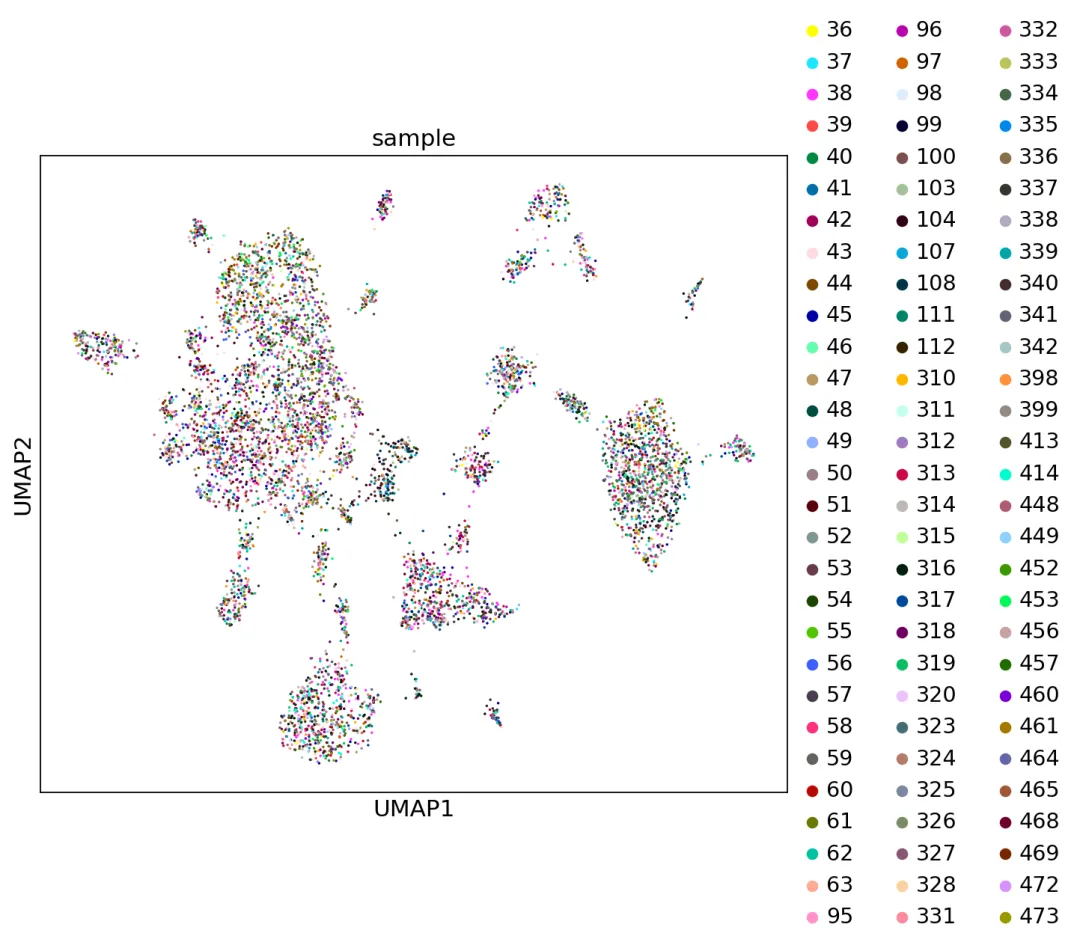
下面是不同组织来源的聚类结果umap:结果也比较均匀
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (8, 7)}): sc.pl.umap(adata, color=["origin"], legend_loc="on data", size=12) 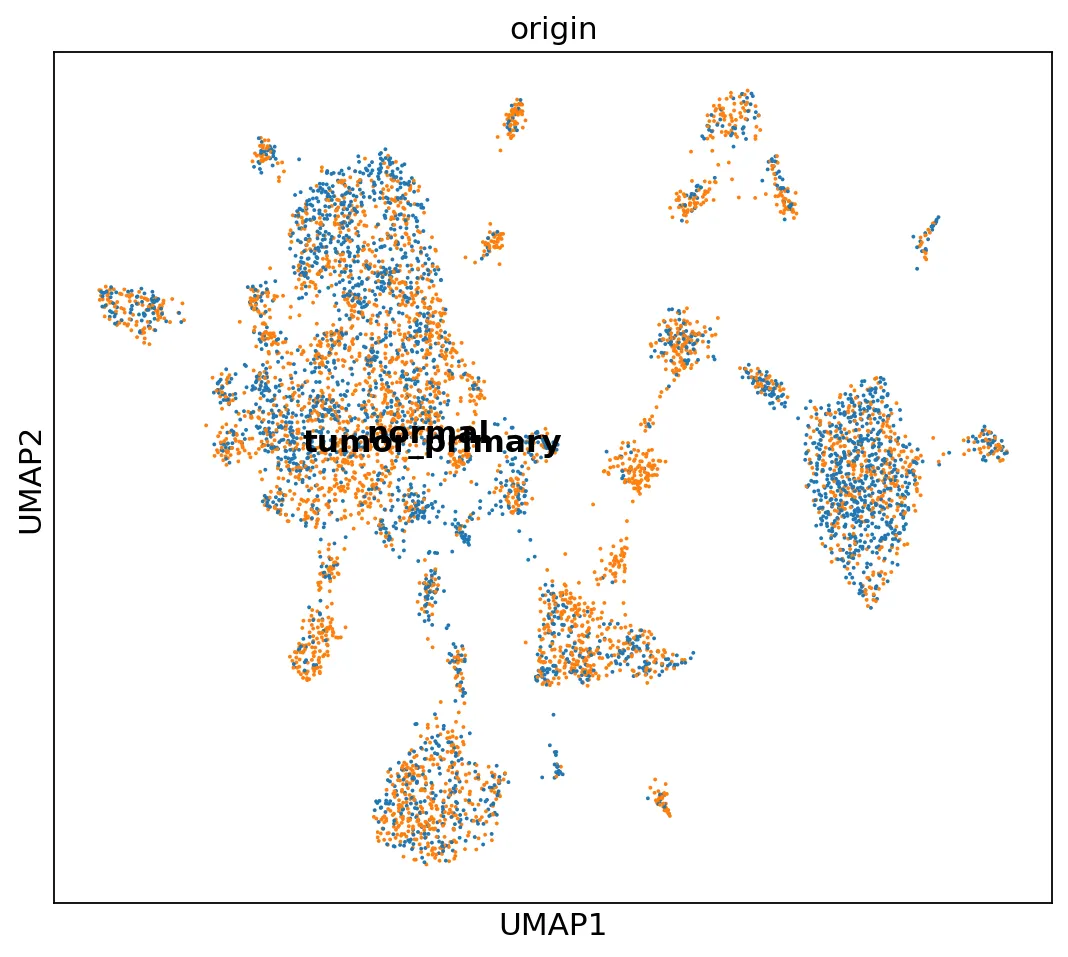
下面是作者的注释信息:可以看到分类结果跟作者的注释信息一致。
# umap图from matplotlib.pyplot import rc_contextwith rc_context({"figure.figsize": (8, 7)}): sc.pl.umap(adata, color=["annot"], legend_loc="on data", size=12) 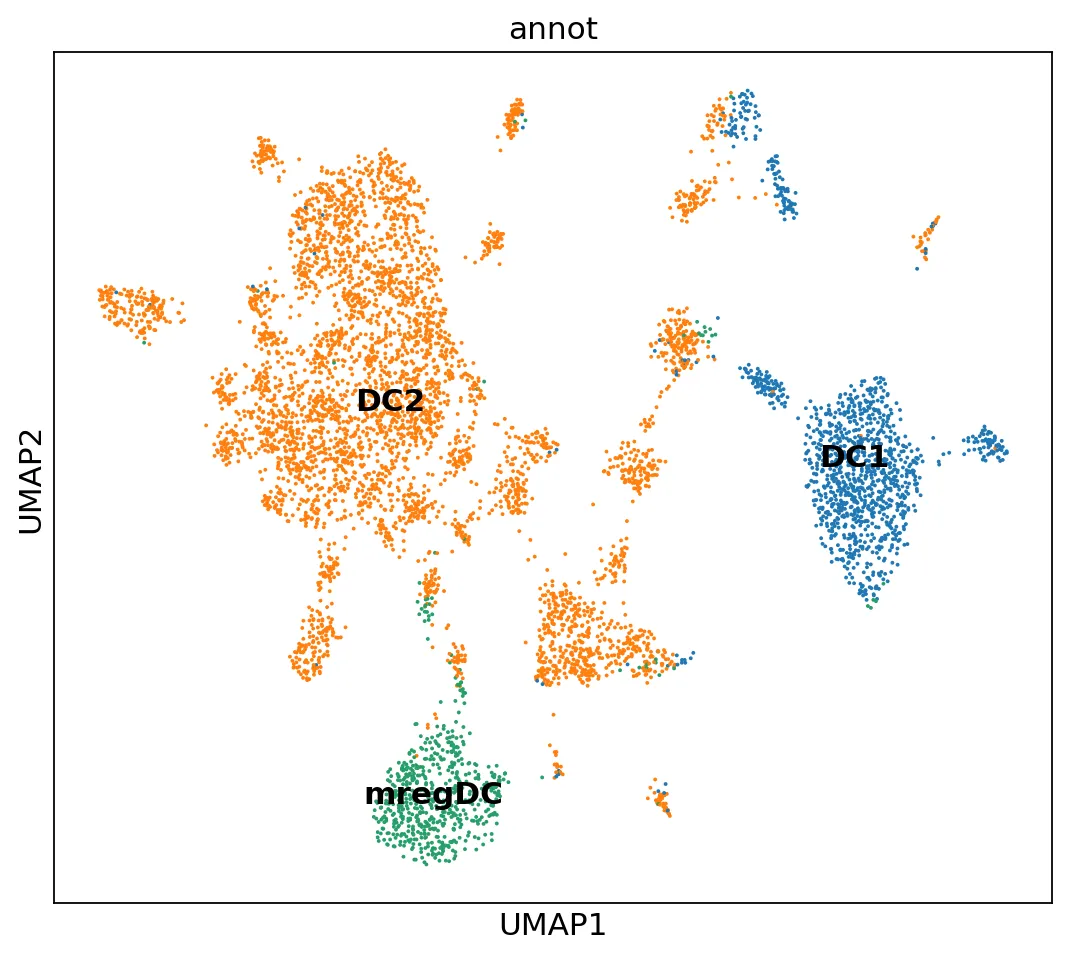
发问:
上面的harmony整合效果如何?
如果你判断不出来,来学习我们这个python单细胞直播系列吧,3月份准备续接起来啦,每周至少一期直播。
添加新叶的微信Biotree123,报暗号python直播学习进群。
今天分享到这里~
友情转发:
生信入门&数据挖掘线上直播课2026年3月班,你的生物信息学入门课 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑

