分享一个ReaxFF-nn 代码:基于GULP/LAMMPS平台的反应式机器学习势函数及其在碳纳米结构热导率计算中的应用。感谢论文的原作者!
================================
在材料模拟与计算化学领域,如何兼顾计算效率与量子力学精度,一直是科学家面临的长期挑战。传统的经验力场虽快但不够准,第一性原理方法虽准但难以处理大体系。
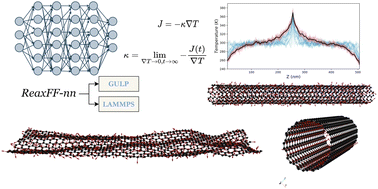
一项发表在 《Physical Chemistry Chemical Physics》 上的研究,巧妙地将机器学习神经网络与经典反应力场相融合,开发出名为 ReaxFF-nn 的机器学习势函数,并成功将其集成到两大主流分子动力学模拟软件 GULP 和 LAMMPS 中。
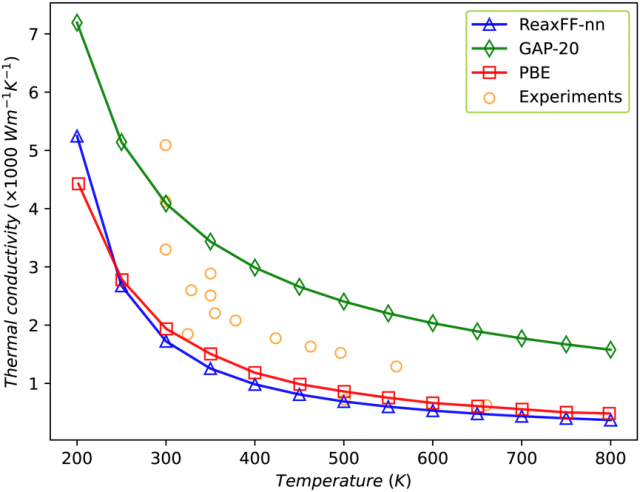
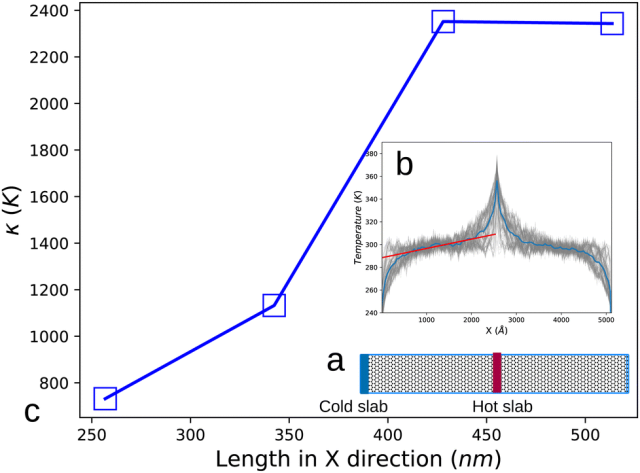
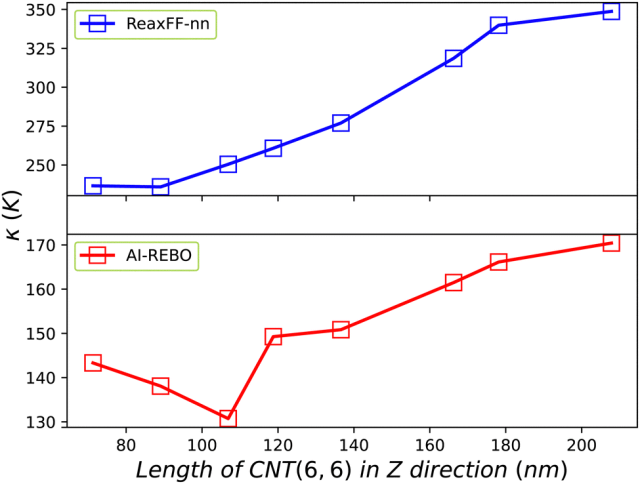
这一成果不仅大幅提升了复杂体系(尤其是化学反应体系)模拟的精度,更在碳纳米材料的热导率计算上展现出与密度泛函理论相媲美的能力。
研究亮点✓ 机器学习与经典力场深度融合✓ 准DFT精度的大规模分子动力学模拟✓ 无缝集成LAMMPS/GULP主流平台✓ 碳材料热导率计算验证优异性能✓ 代码完全开源,推动领域发展
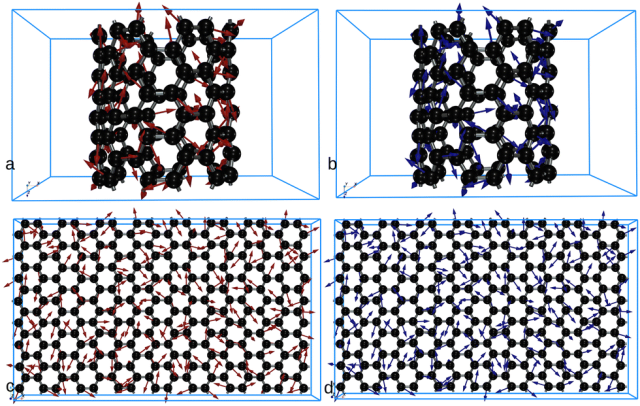
图1. 通过自动微分、GULP和LAMMPS计算的力对比图。(a) 红色箭头表示通过自动微分计算得到的力;(b) 蓝色箭头表示通过我们为碳纳米管实现的GULP代码计算的力;(c) 红色箭头表示通过自动微分计算得到的力;(d) 蓝色箭头表示通过我们为石墨烯片实现的LAMMPS代码计算的力。
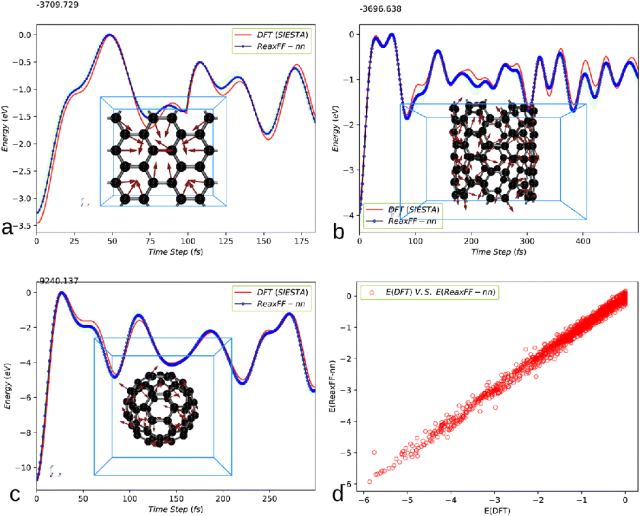
图2. DFT数据与ReaxFF-nn预测结果的对比图。DFT-MD数据通过SIESTA软件采集,采用PBE泛函和双ζ极化基组。图中分别展示了(a)石墨烯、(b) 6×6碳纳米管和(c)富勒烯(C₆₀)的对比结果。(d) 展示了ReaxFF-nn对所有训练数据的能量误差评估指标。
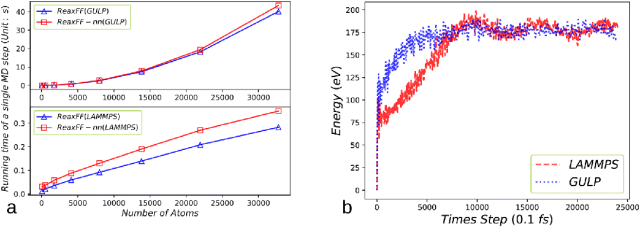
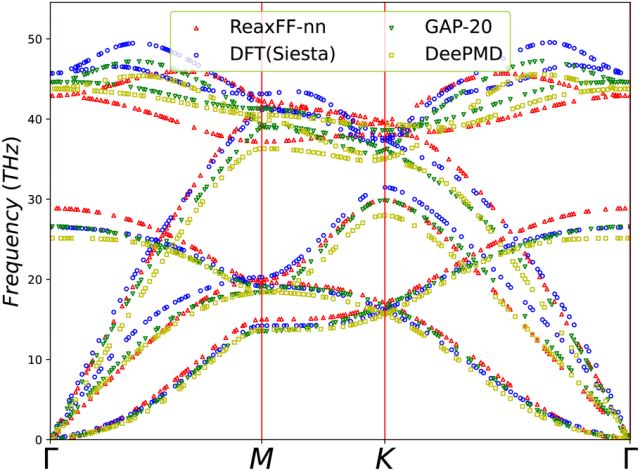
图3. (a) 单个MD步长的耗时对比:ReaxFF-nn与基于GULP包(上图)和LAMMPS包(下图)的ReaxFF在不同原子数下的计算时间对比。(b) 在长达2.5 ps的分子动力学模拟中,使用GULP和LAMMPS包模拟10 × 10 × 10石墨烯片层势能随时间的变化情况。图4. 二维石墨烯晶格层声子色散关系的对比:GAP-20、DeePMD与ReaxFF-nn势函数的计算结果。声子色散关系通过Phonopy软件包计算得出。
================================
以上是我们分享的一些经验或者文章的搬运,或有不足,欢迎大家指出。若留言未回复,重要的消息可以留言再提醒一下。
如有侵权,请联系我们立马删除!
👇
文章题目:
ReaxFF-nn: a reactive machine-learning potential in GULP/LAMMPS and its applications in the thermal conductivity calculations of carbon nanostructures
https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp00535j
👇