python版单细胞注释:自动注释之celltypist
- 2026-07-06 20:31:15
前面看到一个学妹在学习python,她的一篇推文中提到了一个python代码的学习资源,今天来看看里面的细胞注释部分。
https://www.sc-best-practices.org/preamble.html
昨天我们看了细胞手动注释部分:python版单细胞注释:基于已知marker基因注释
自动注释方法基于不同原理,有时需要预定义的标记基因集,其他时候则在已有的完整单细胞RNA测序数据集上进行训练,由此产生的标注质量可能参差不齐。因此,将这些自动注释方法视为标注过程的起点而非终点非常重要。关于自动化标注方法的更详细讨论,还可参考多篇综述:
[Pasquini et al., 2021]:Automated methods for cell type annotation on scrna-seq data. Computational and Structural Biotechnology Journal [Abdelaal et al., 2019]:A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biology
自动化生成标注的质量可能参差不齐。标注质量取决于:
所选分类器的类型:先前基准研究表明,不同类型分类器的表现通常不相上下。
分类器训练数据的质量:如果训练数据标注不完善或分辨率较低或其标注存在噪声,分类器也会呈现相同问题。
用户数据与分类器训练数据的相似度:例如,若分类器基于drop-seq单细胞数据集训练,而用户数据是10X单核而非单细胞drop-seq数据,则可能降低标注质量。相比在单一数据集上训练的分类器,基于包含多样化数据集的跨数据集图谱训练的分类器可能提供更稳健、更优质的标注。
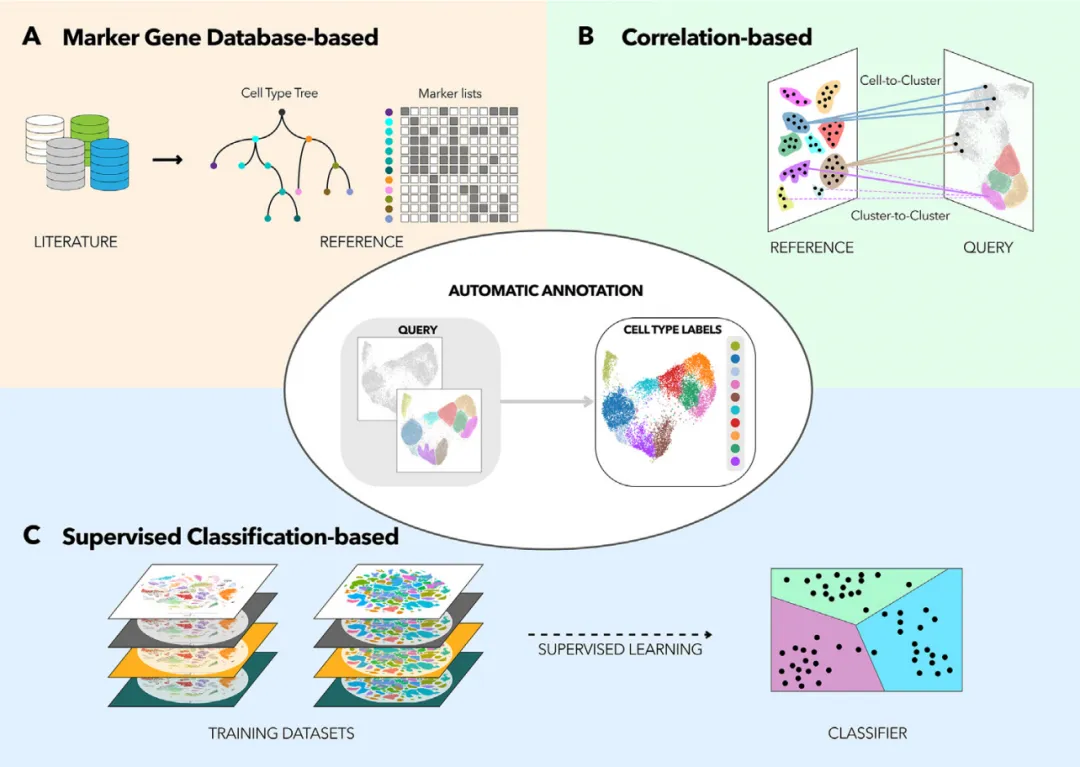
基于标记基因的分类器
有一类自动化细胞类型标注方法依赖于预定义的标记基因集。细胞根据这些标记基因的表达水平被分类为特定细胞类型。此类方法的典型代表包括 Garnett [Pliner et al., 2019] 和 CellAssign [Zhang et al., 2019]。这些模型所依据的标记基因集越稳健、普适性越强,模型性能就越好。
这类方法的一个优势在于其更高的可解释性:我们能够明确知晓分类所依据的具体基因。
Garnett:Published: 09 September 2019,文献标题为《Supervised classification enables rapid annotation of cell atlases》. Nature Methods。
这个方法我看了软件地址,目前已经停止更新很久了,放弃。
https://github.com/cole-trapnell-lab/garnett/
CellAssign:09 September 2019 ,标题为《Probabilistic cell-type assignment of single-cell rna-seq for tumor microenvironment profiling》. Nature Methods,
CellAssign同样已经停止更新很久,放弃。
https://github.com/Irrationone/cellassign
基于更广泛基因集的分类器
另一种方案是采用基于更广泛基因集(数千甚至更多基因)的分类器,从而更充分地利用单细胞RNA测序数据的广度。这类分类器通过先前已标注的数据集或细胞图谱进行训练,典型代表包括CellTypist [Conde et al., 2022]。
尝试CellTypis:
https://www.celltypist.org
1.数据预处理
数据见:python版单细胞注释:基于已知marker基因注释
根据CellTypist教程的要求,我们需要将数据预处理为每个细胞计数标准化至10,000后,再进行log1p转换:
adata_celltypist = adata.copy() # make a copy of our adataadata_celltypist.X = adata.layers["counts"] # set adata.X to raw countssc.pp.normalize_total(adata_celltypist, target_sum=10**4) # normalize to 10,000 counts per cellsc.pp.log1p(adata_celltypist) # log-transform# make .X dense instead of sparse, for compatibility with celltypist:adata_celltypist.X = adata_celltypist.X.toarray()2.下载celltypist模型
可以同时尝试"Immune_All_Low"和"Immune_All_High"这两种模型(它们分别以更精细的低分辨率级别和更粗略的高分辨率级别来标注免疫细胞类型)。
下载的结果保存在这里:/home/zhangj/.celltypist/data/models
models.download_models( force_update=True, model=["Immune_All_Low.pkl", "Immune_All_High.pkl"])model_low = models.Model.load(model="Immune_All_Low.pkl")model_high = models.Model.load(model="Immune_All_High.pkl")# 针对每个模型,我们可以查看其包含的细胞类型,以确认是否涵盖骨髓细胞类型:model_high.cell_typesmodel_low.cell_types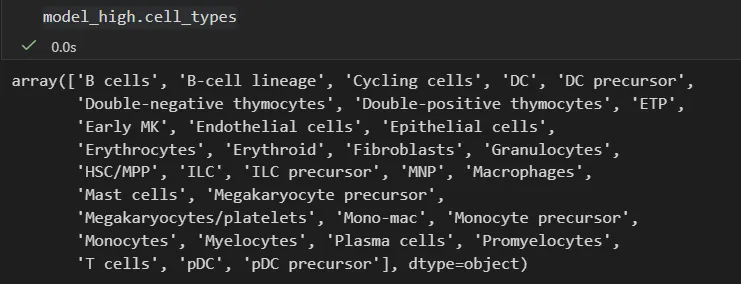
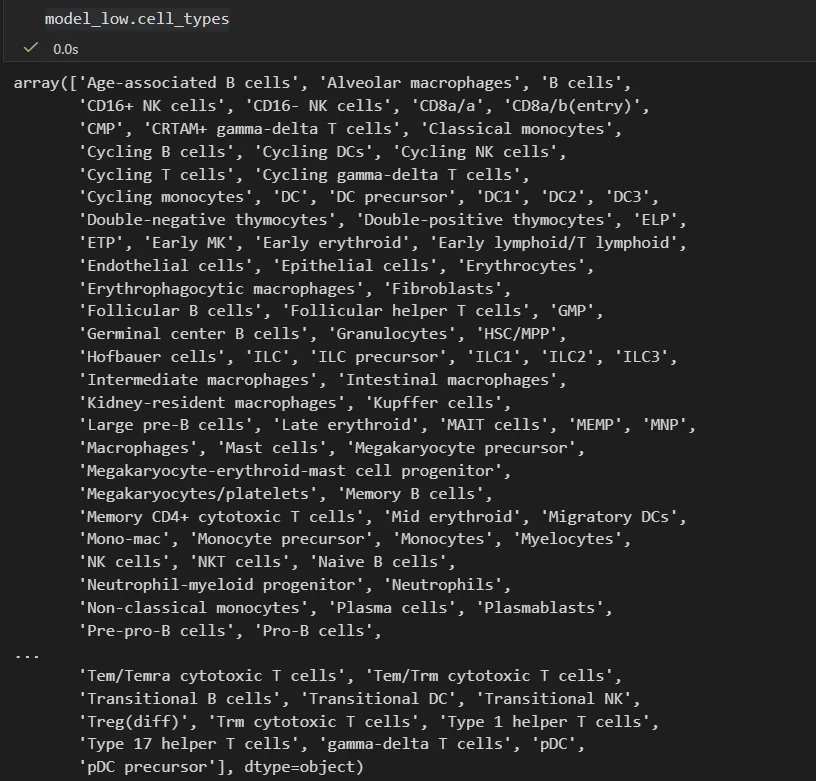
这些模型包含了许多不同类型的免疫细胞祖细胞!
3.运行粗略分类模型
首先运行粗略分类模型:
predictions_high = celltypist.annotate( adata_celltypist, model=model_high, majority_voting=True)# 将预测结果转换为adata格式以获取完整输出predictions_high_adata = predictions_high.to_adata()# 将结果复制到我们原始的AnnData对象中:adata.obs["celltypist_cell_label_coarse"] = predictions_high_adata.obs.loc[ adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_coarse"] = predictions_high_adata.obs.loc[ adata.obs.index, "conf_score"]4.运行更精细分类模型
现在对更精细的注释进行相同操作:
# 精细注释predictions_low = celltypist.annotate( adata_celltypist, model=model_low, majority_voting=True)predictions_low_adata = predictions_low.to_adata()adata.obs["celltypist_cell_label_fine"] = predictions_low_adata.obs.loc[ adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_fine"] = predictions_low_adata.obs.loc[ adata.obs.index, "conf_score"]5.看看预测结果
将注释结果放到umap上面看看:
# 绘图看看那预测结果,粗注释sc.pl.umap( adata, color=["celltypist_cell_label_coarse", "celltypist_conf_score_coarse"], frameon=False, sort_order=False, wspace=1,)
详细注释:
sc.pl.umap( adata, color=["celltypist_cell_label_fine", "celltypist_conf_score_fine"], frameon=False, sort_order=False, wspace=1,)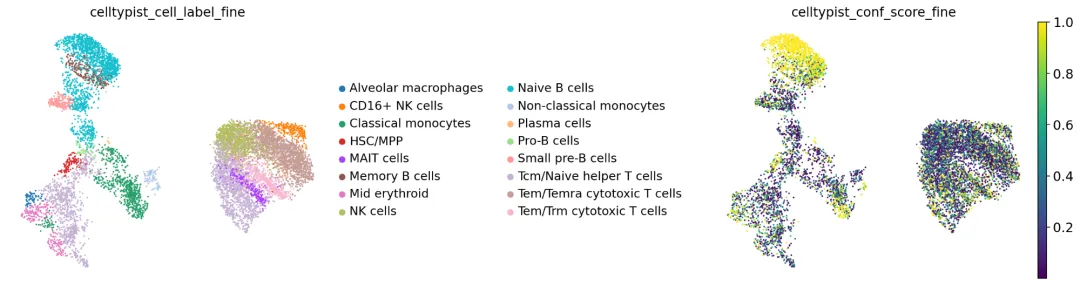
6.评估预测结果
评估这些注释质量的一种方法是观察识别的细胞类型相似性是否符合我们的预期:
sc.pl.dendrogram(adata, groupby="celltypist_cell_label_fine")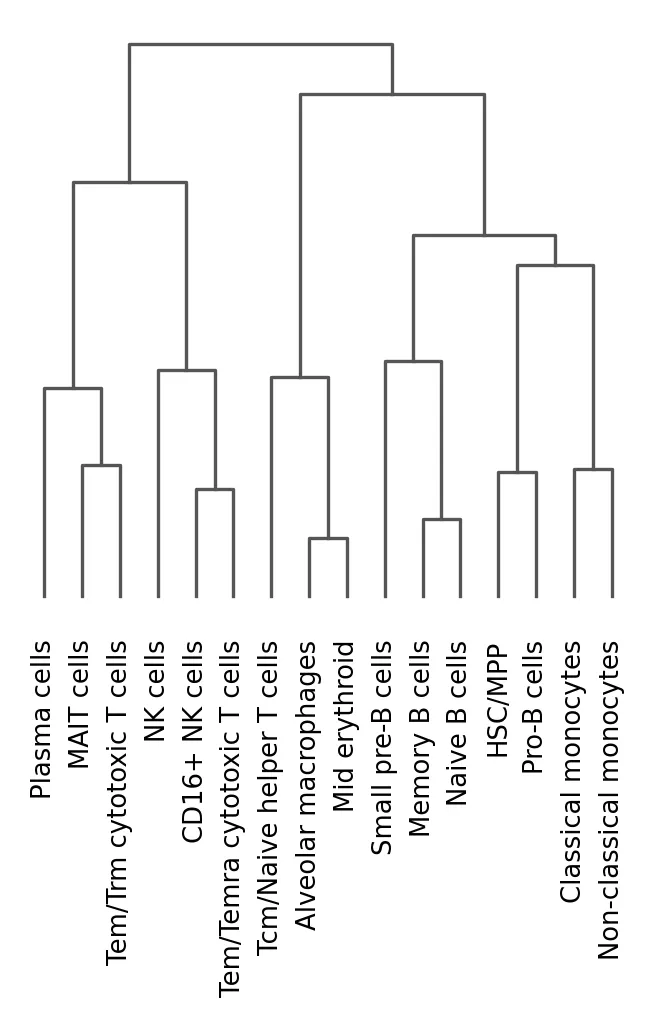
这个树状图部分反映:
根据细胞类型关系的先验知识,看到例如B细胞基本聚集在一起,但也观察到一些意外模式:Tcm/Naive辅助T细胞与红细胞和巨噬细胞聚类,而非其他T细胞。这是一个危险信号!可能Tcm/Naive辅助T细胞的注释是错误的。
查看一下之前的手动注释:
sc.pl.umap( adata, color=["manual_celltype_annotation"], frameon=False,)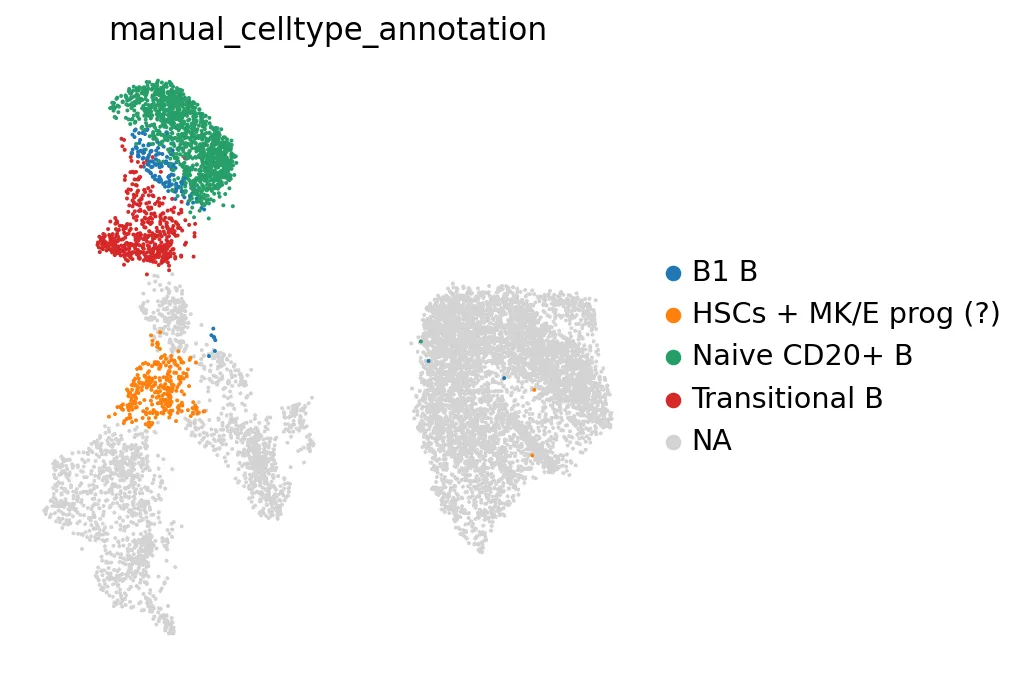
可以看到,之前手动标注的初始B细胞与部分自动标注的初始B细胞吻合良好。同样,称为过渡B细胞的部分在自动标注中被称为“小前B细胞”,而手动标注的B1 B细胞对应他们的记忆B细胞!
但是,在手动标注的HSC + MK/E祖细胞簇中,自动精细注释显示为T细胞与HSC/多能祖细胞的混合,表明这两种注释方法存在部分矛盾。
如果观察自动注释的置信度得分,可以看到大部分细胞的标注置信度相对较低,这明确提示:
自动注释不能未经仔细验证和人工审阅就直接采用这些标注!
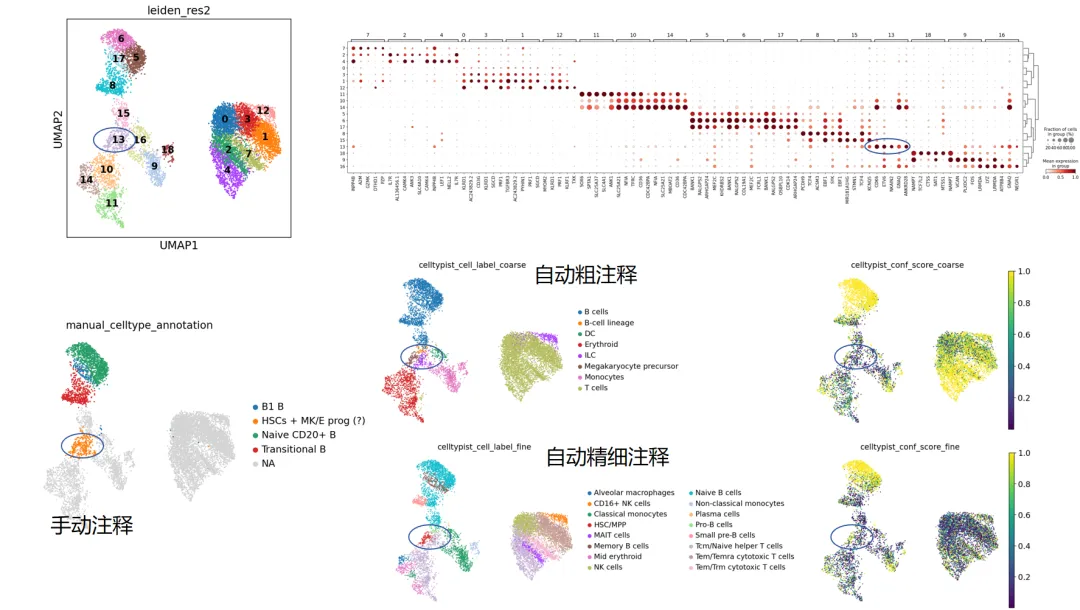
总结
以上结果强调了应谨慎使用自动标注算法,并应将其视为数据标注的起点,而非最终结果。归根结底,已知标记基因的表达仍是细胞类型标注最受认可的依据。
转发:
生信入门&数据挖掘线上直播课2026年1月班,你的生物信息学入门课 时隔5年,我们的生信技能树VIP学徒继续招生啦 满足你生信分析计算需求的低价解决方案 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 【善假AI】随机位移迭代代码生成(用于修正化学式元素交叉大表118*118)
- 多线程编程中,你以为加了锁就安全了吗?
- 2026年编程新姿势:手把手教你把 GLM-4.7 接入 Claude Code,这波羊毛必须薅!
- 人工智能OPC夜校首期开讲!借AI无代码编程解锁OPC创业
- SQL编程大赛第4名马尾蛟:一条 SQL 解数独?PostgreSQL 递归 CTE 封神思路全拆解
- PHP打包部署的最佳实践
- PHP函数mysqli_num_rows():获取结果集中的行数
- 央视报道:核桃编程AI编程课走进全国中小学!核桃编程创始人曾鹏轩再登CCTV
- 小白完全不懂技术,AI编程怎么学?尝试从这4点入手��️
- Meta 产品经理:零代码背景,如何用AI工具独立交付产品?丨Lenny's Podcast
